Open Journal of Anesthesiology
Vol.3 No.3(2013), Article ID:31256,4 pages DOI:10.4236/ojanes.2013.33035
Pheochromocytoma Anesthetic Management
![]()
1Resident in Anesthesiology, Santa Casa de Misericordia de Sao Paulo, Sao Paulo, Brazil; 2Anesthesiologist, Santa Casa de Misericordia de Sao Paulo, Sao Paulo, Brazil; 3Director of Pain Medicine, Santa Casa de Misericordia de Sao Paulo, Sao Paulo, Brazil; 4Director of Anesthesiology Service, Santa Casa de Misericordia de Sao Paulo, Sao Paulo, Brazil.
Email: *dandonkim@gmail.com
Copyright © 2013 Daniel D. Kim et al. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Received February 13th, 2013; revised March 30th, 2013; accepted April 15th, 2013
Keywords: Pheochromocytoma; Anesthesia Management
ABSTRACT
Pheochromocytomas are catecholamine producing tumors and although uncommon present a great challenge to the anesthesiologist since it has nonspecific clinical symptoms and risk of critical events, including death when not previously diagnosed. Clinical manifestation is variable, unspecific and depends on the catecholamine production profile. The classic triad of headache, palpitation and diaphoresis is present in up to 70% of the cases and only 50% have sustained hypertension. The best approach for pheochromocytoma treatment is surgical excision of the affected adrenal gland. The introduction of alpha adrenergic blockade medication, such as phentolamine and phenoxybenzamine had the highest impact in perioperative mortality reduction due to inhibition of the deleterious effect of vasoconstriction. The majority of anesthetic techniques and drugs are considered safe. Post-operative care in intensive care unit is advisable since patients may present instability of blood pressure and hypoglycemia. Genetic testing should be done in first-degree relatives of confirmed cases or when a genetic syndrome is suspected.
1. Introduction
Pheochromocytomas are catecholamine producing tumor originated from chromaffin cells and up to 80% are located in the adrenal gland. Although uncommon they present a great challenge to the anesthesiologist since it has unspecific clinical symptoms, complex and not widely available diagnosis testing and risk of critical events, including death when not diagnosed. New research has found that up to 30% of pheochromocytoma cases are associated with autosomal genetic mutation [1,2].
2. Objectives
The proposal of this article is review the recent literature about clinical manifestation, diagnosis criteria, treatment options and anesthetic management of pheochromocytoma.
3. Method
For the review, scientific articles published between 2000 and 2012 in the PubMed Central (PMC-NCBI) databank where selected, preferably review and meta-analysis written in English containing the MeSH terms pheochromocytoma, phaechromocytoma, anesthesia and anaesthesia. Case report, experimental studies and uncommon presentations were excluded.
4. Results
The search retrieved 124 original articles respecting the inclusion and exclusion criteria described. From the original research 18 articles where selected for this review.
4.1. Clinical Manifestation and Diagnosis
The clinical manifestation is variable, unspecific and depends on the catecholamine production profile. The classic triad of headache, palpitation and diaphoresis is present in up to 70% of the cases and only 50% have sustained hypertension. The annual incidence is estimated in 2 to 8 cases per million in general population and prevalence of 0.1% to 0.6% in hypertensive population [3,4].
The best exam for diagnosis is the measurement of plasmatic metanephrines (Sensitivity of 99% and specificity of 89%) and since many medications can influence this test (Table 1) proper patient preparation is of upmost importance. The best option for topographic location is the use of catecholamine synthesis metabolic marker such as scintigraphy with 123I-MIBG and PET scan with 18Ffluorodopamine [3,4].
4.2. Treatment Options
The best approach for pheochromocytoma treatment is the surgical excision of the affected adrenal gland. Due to retroperitoneal anatomic positioning the preferably technique is laparoscopy [4].
For metastatic disease, there are other options like symptomatic treatment with adrenergic blockers, radiotherapy with 133I-MIBG, chemotherapy and conventional radiotherapy. The prognosis of this presentation is poor with less than 50% survival in 5 years [1,4].
4.3. Anesthetic Management
An optimal pre-anesthetic management is vital for pheochromocytoma treatment and the goal is clinical control of the disease with reduction of plasmatic catecholamine levels. The introduction of alpha adrenergic blockade medication (Table 2), such as phentolamine and phenoxybenzamine had the highest impact in perioperative mortality reduction because it inhibits the deleterious effect of vasoconstriction [5-7].
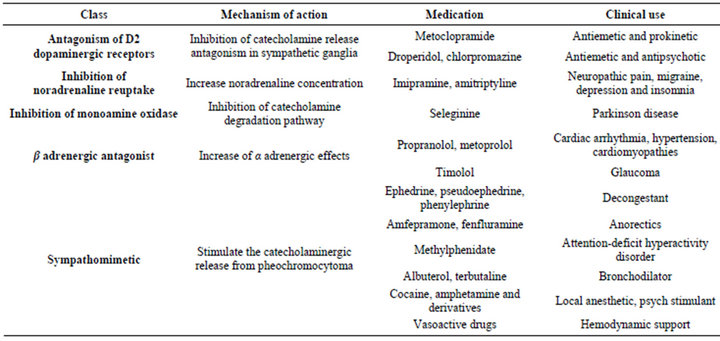
Table 1. Medications that influence laboratory testing or treatment of pheochromocytoma.
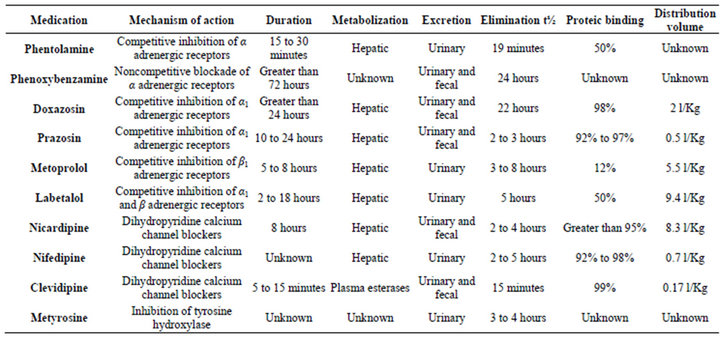
Table 2. Mechanism of action and pharmacokinetics of medications used for pheochromocytoma control.
During surgery the objective is maintenance of hemodynamic stability, since normal surgical stimuli (patient positioning, anesthesia induction, intubation, and tumor manipulation) may cause severe hypertension. The majority of anesthetic techniques and drugs are considered safe. Along with regular surgical monitoring, invasive blood pressure measurement is highly recommended [7- 13].
After tumor excision hypotension may begin due to residual effect of alpha adrenergic antagonists, increase in venous capacitance and intraoperative bleeding, but is usually manageable leading to a near zero perioperative mortality in recent studies [14-17].
4.4. Post-Operative Management
Is advisable the post-operative care in intensive care unit since many patients may present blood pressure instability (up to 50% may have hypertension and increase of plasmatic levels of catecholamine) and hypoglycemia (patients may have a decreased glycogen level due to the increase in glycogenolysis and lipolysis caused by adrenergic stimulus) [18-21].
Lifelong follow up with plasmatic dosage of catecholamine and metanephrine is recommended since there are cases of late tumoral relapse in literature. Genetic testing should be done in first-degree relatives of confirmed cases or when genetic syndrome is suspected (café au lait spots, cerebellar tumor, thyroid medullar carcinoma, hyperparathyroidism) [8,9].
5. Discussion
Although uncommon type of tumor, pheochromocytomas present a great challenge to the anesthesiologist, since it has unspecific clinical symptoms, complex detection tests and possibility of unfavorable results, including death when not previously diagnosed.
Familiar or personal history of critical perioperative events associated with a detailed physical examination during pre-anesthetic evaluation are the main tools for the suspicion of these tumors.Clinical control with alpha adrenergic antagonistmedication had the highest impact in reduction of perioperative mortality and the majority of the experts consider adequate 14 to 21 days of treatment before surgery.
During surgery hemodynamic instability may arise due to nociceptive stimulus or tumoral manipulation, therefore hypotensive medications should be ready before anesthetic induction (Table 3). Commonly symptomatic adjuvant medication used in anesthesia (metoclopramide, droperidol) should be judiciously selected since it may trigger adrenergic crisis (Table 1). Postoperative care in an intensive care unit is recommended.
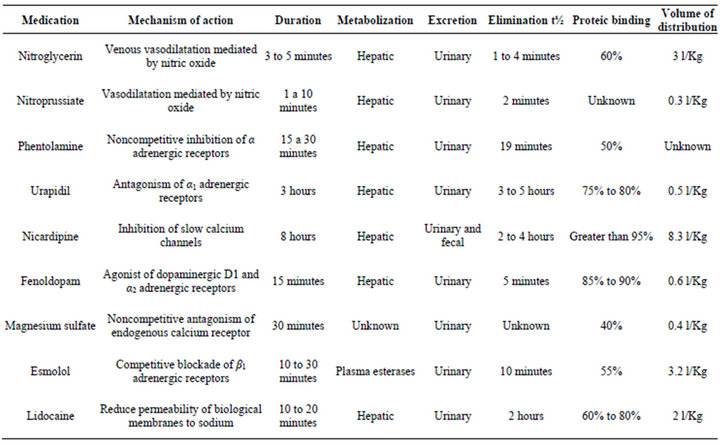
Table 3. Mechanism of action and pharmacokinetics of medications used for acute blood pressure control in pheochromocytoma.
All patients with pheochromocytoma should have lifelong follow up since late relapse have been reported. Relatives of confirmed pheochrmocytomas cases should receive information about the increased risk of cathecolaminergic crisis.
REFERENCES
- H. Chen, R. S. Sippel, M. S. O’Dorisio, A. I. Vinik, R. V. Lloyd and K. Pacak, “North American Neuroendocrine Tumor Society (NANETS). The North American Neuroendocrine Tumor Society Consensus Guideline for the Diagnosis and Management of Neuroendocrine Tumors: Pheochromocytoma, Paraganglioma, and Medullary Thyroid Cancer,” Pancreas, Vol. 39, 2010, pp. 775-783. doi:10.1097/MPA.0b013e3181ebb4f0
- K. Pacak, G. Eisenhofer, H. Ahlman, S. R. Bornstein, A. P. Gimenez-Roqueplo, A. B. Grossman, N. Kimura, M. Mannelli, A. M. McNicol and A. S. Tischler, “International Symposium on Pheochromocytoma. Pheochromocytoma: Recommendations for Clinical Practice from the First International Symposium. October 2005,” Nature Clinical Practice Endocrinology & Metabolism, Vol. 3, 2007, pp. 92-102. doi:10.1038/ncpendmet0396
- W. F. Young, Williams Textbook of Endocrinology, Saunders/Elsevier, Philadelphia, 2008.
- J. W. Lenders, G. Eisenhofer, M. Mannelli and K. Pacak, “Phaeochromocytoma,” Lancet, Vol. 20-26, No. 366, 2005, pp. 665-675. doi:10.1016/S0140-6736(05)67139-5
- S. S. Bajwa and S. K. Bajwa, “Implications and Considerations during Pheochromocytoma Resection: A Challenge to the Anesthesiologist,” Indian Journal of Endocrinology & Metabolism, Vol. 15, No. S4, 2011, pp. S337-S344. doi:10.4103/2230-8210.86977
- G. Eisenhofer, G. Rivers, A. L. Rosas, Z. Quezado, W. M. Manger and K. Pacak, “Adverse Drug Reactions in Patients with Phaeochromocytoma: Incidence, Prevention and Management,” Drug Safety, Vol. 30, 2007, pp. 1031-1062. doi:10.2165/00002018-200730110-00004
- M. A. Kinney, B. J. Narr and M. A. Warner, “Perioperative Management of Pheochromocytoma,” Journal of Cardiothoracic and Vascular Anesthesia, Vol. 16, 2002, pp. 359-369. doi:10.1053/jcan.2002.124150
- K. Pacak, “Preoperative Management of the Pheochromocytoma Patient,” Indian Journal of Endocrinology & Metabolism, Vol. 92, 2007, pp. 4069-4079. doi:10.1210/jc.2007-1720
- C. Lentschener, S. Gaujoux, A. Tesniere and B. Dousset, “Point of Controversy: Perioperative Care of Patients Undergoing Pheochromocytoma Removal-Time for a Reappraisal?” European Journal of Endocrinology, Vol. 165, 2011, pp. 365-373. doi:10.1530/EJE-11-0162
- R. T. Wall, “Stoelting’s Anesthesia and Co-Existing Disease,” Saunders/Elsevier, Philadelphia, 2008. doi:10.1016/B978-1-4160-3998-3.10016-8
- I. Jugovac, M. Antapli and S. Markan, “Anesthesia and Pheochromocytoma,” International Anesthesiology Clinics, Vol. 49, 2011, pp. 57-61. doi:10.1097/AIA.0b013e3181ff4db0
- T. C. Westfall and D. P. Westfall, “The Pharmacologycal Basis of Therapeutics,” McGraw Hill, New York, 2011.
- H. Bruynzeel, R. A. Feelders, T. H. Groenland, A. H. van den Meiracker, C. H. van Eijck, J. F. Lange, W. W. de Herder and G. Kazemier, “Risk Factors for Hemodynamic Instability during Surgery for Pheochromocytoma,” Journal of Clinical Endocrinology & Metabolism, Vol. 95, 2010, pp. 678-685. doi:10.1210/jc.2009-1051
- A. Nizamoğlu, Z. Salihoğlu and M. Bolayrl, “Effects of Epidural-and-General Anesthesia Combined versus General Anesthesia during Laparoscopic Adrenalectomy,” Surgical Laparoscopy Endoscopy & Percutaneous Techniques, Vol. 21, 2011, pp. 372-379. doi:10.1097/SLE.0b013e31822dd5e1
- T. Isosu, S. Obara, S. Ohashi, A. Hosono, Y. Nakano, T. Imaizumi, M. Mogami, H. Iida and M. Murakawa, “Examination of the Usefulness of Non-Invasive Stroke Volume Variation Monitoring for Adjusting Fluid Supplementation during Laparoscopic Adrenalectomy in Patients with Pheochromocytoma,” Fukushima Journal of Medical Science, Vol. 58, 2012, pp. 78-81. doi:10.5387/fms.58.78
- R. Domia and H. Sulaa, “Pheochromocytoma, the Challenge to Anesthesiologists,” Journal of Endocrinology & Metabolism, Vol. 1, 2011, pp. 97-100.
- M. A. Kinney, M. E. Warner, J. A. van Heerden, T. T. Horlocker, W. F. Young Jr., D. R. Schroeder, P. M. Maxson and M. A. Warner, “Perianesthetic Risks and Outcomes of Pheochromocytoma and Paraganglioma Resection,” Anesthesia & Analgesia, Vol. 91, 2000, pp. 1118-1123.
- M. F. James and L. Cronjé, “Pheochromocytoma Crisis: The Use of Magnesium Sulfate,” Anesthesia & Analgesia, Vol. 99, 2004, pp. 680-686. doi:10.1213/01.ANE.0000133136.01381.52
- S. Herroeder, M. E. Schönherr, S. G. De Hert and M. W. Hollmann, “Magnesium—Essentials for Anesthesiologists,” Anesthesiology, Vol. 114, 2011, pp. 971-993. doi:10.1097/ALN.0b013e318210483d
- M. S. Lord and J. G. Augoustides, “Perioperative Management of Pheochromocytoma: Focus on Magnesium, Clevidipine, and Vasopressin,” Journal of Cardiothoracic and Vascular Anesthesia, Vol. 26, 2012, pp. 526-531. doi:10.1053/j.jvca.2012.01.002
- A. N. Van der Horst-Schrivers, M. N. Kerstens and B. H. Wolffenbuttel, “Preoperative Pharmacological Management of Phaeochromocytoma,” Netherlands Journal of Medicine, Vol. 64, 2006, pp. 290-295.
NOTES
*Corresponding author.