Open Journal of Molecular and Integrative Physiology
Vol.3 No.2(2013), Article ID:31932,8 pages DOI:10.4236/ojmip.2013.32014
The phka1 deficient I/LnJ mouse exhibits endurance exercise deficiency with no compensatory changes in glycolytic gene expression
![]()
Department of Biology, Western Kentucky University, Bowling Green, USA
Email: *Nancy.rice@wku.edu
Copyright © 2013 Ashley M. Mefford et al. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Received 30 March 2013; revised 30 April 2013; accepted 7 May 2013
Keywords: Phosphorylase Kinase; Glycogen Storage Disease; Exercise; Skeletal Muscle; I/LnJ
ABSTRACT
During exercise, phosphorylase kinase (PhK) is the key regulatory enzyme responsible for maintaining glycogenolytic flux to sustain muscle contraction. The absence of PhK in skeletal muscle results in glycogen storage disease (GSD) Type IX which is characterized by muscle weakness and rapid fatigue upon exercise. In this study, we have used the phka1 deficient I/LnJ mouse model of GSD to investigate the physiological and genetic adaptations that occur in response to voluntary exercise. When quantified over training periods of either 1, 2, or 5 weeks, I/LnJ mice ran significantly less time/day and distance/day than agematched C57/Bl6 mice. Cumulatively after five weeks, adult I/LnJ mice ran ~1/2 the total time and distance of wild-type mice, 116 ± 6 hours and 211 ± 23 km versus 194 ± 3 hours and 418 ± 4 km, respectively. After 5 weeks, C57/Bl6 mice demonstrated an increase in endurance as a result of aerobic training; this observed physiological adaptation was not present in I/LnJ mice. The decrease in total distance run by I/LnJ mice was not due to a reduction in speed; juvenile and adult I/LnJ mice ran ~75% - 80% as fast as C57/Bl6 mice. When transcription of glycolytic genes glucose transporter 4 (scla1), pyruvate dehydrogenase (pdha1), and phosphofructokinase (pfk) were quantified at the end of each training period, no significant differences in expression levels were found between mouse strains, suggesting that non-glycolytic mechanisms work to maintain the muscle function observed in the I/LnJ mice.
1. INTRODUCTION
During exercise, the glucose polymer glycogen serves as the primary fuel for sustaining muscle contraction. Glycogen is hydrolyzed via the cascade activation of glycolgenolysis as a result of conversion of glycogen phosphorylase, Pb, to its activated form, Pa, by the key regulatory enzyme phosphorylase kinase (PhK) [1,2]. Structurally, PhK is a hexadecameric enzyme with a stoichiometry of (αβγδ)4 where α and β are regulatory, γ is catalytic, and δ, is an intrinsic calmodulin molecule. Functionally, PhK sits at the interface between cellular signaling and metabolic activation, and integrates neural (Ca2+), hormonal (cAMP) and metabolic (ADP) stimuli in order to modulate glycogenolytic flux in skeletal muscle for ATP production [3,4].
In humans, glycogen storage diseases (GSDs) can result from a deficiency in any of the enzymes involved in either the synthesis or degradation of glycogen. GSD Type IX results from a deficiency in PhK, and there is significant heterogeneity in the disease as it can result from mutations in any of the seven genes that encode for PhK subunits [5]. In particular for this study, humans that have mutations in PHKA1, the gene encoding the muscle-specific variant of the α subunit, present with muscle weakness and rapid fatigue upon exercise [6,7], as well as muscle cramps and stiffness combined with exercise intolerance [8-10]. A naturally occurring inbred mouse strain, I/LnJ, also has a mutation in phka1 which results in an almost complete loss of PhK activity, 0.2% - 0.3% of normal levels in the limb and back muscles and 1.5% of normal amounts in the soleus muscle [11-16], as well as the absence of Pa [17,18]. While less efficient, these mice can degrade glycogen in isolated tetanically contracting muscle [19].
In addition to muscle glycogen stores, the peripheral blood is also a source of glucose for skeletal muscle during exercise. Uptake of glucose occurs via glucose transporter 4 (GLUT4) [20,21] which is translocated to the plasma membrane in response to contractile activity [22]. Studies have shown that increases in GLUT4 mRNA and protein expression can occur as both an early and a longterm adaptive response to exercise, with early increases in GLUT4 expression being advantageous for prolonged, low intensity exercise [23-25]. Furthermore, increases in other glycolytic regulatory enzymes, such as phosphofructokinase, increase in response to exercise as well [26,27].
Thus, in this study we evaluated the I/LnJ mouse strain as a model for human GSD Type IX, by comparing its exercise capacity for voluntary free wheel running to normal wild type mice, and determining whether or not, as a result of a deficiency in muscle PhK, it expresses compensatory changes in glucose transport or glycolytic gene expression.
2. METHODS
2.1. Mice and Voluntary Wheel Running
All protocols used in this study were conducted with the approval of the Institutional Animal Care and Use Committee (IACUC) at Western Kentucky University. Approximately 100 total wild type C57/Bl6 and the PhK deficient I/LnJ mice (also referred to as I-strain) were used for all experiments. Mice were housed in an IACUC approved facility with water and food provided ad libitum. For any given litter, mice were randomly assigned to a particular experimental group or to non-exercised litter mate control group. Adult and juvenile mice, 12 - 14 weeks and 6 - 8 weeks of age, respectively, were placed in individual cages (47 × 26 × 14.5 cm) with a standard 6 inch hamster wheel equipped with a SigmaTM speedometer attached to each wheel. The counter was equipped with a digital magnet that is activated by wheel rotation. Two different development ages were chosen to evaluate since it is known, at least for liver-specific GSD IX that results from mutations in PHKA2, that disease phenotypes often regress with age. Human muscle-specific GSD IX is relatively rare, and therefore it is not known if such developmental changes also occur in this disorder.
Wheels were calibrated as per manufacturer’s instructtions based upon wheel dimensions. Mice were allowed to voluntarily exercise on the wheels for either 1, 2, or 5 weeks. Voluntary wheel running is a valid exercise paradigm to assess physiological adaptations in mice and is advantageous over other non-voluntary methods in that it is inexpensive and produces less stress to the animals by allowing them to exercise when they want, to the extent they want, with no handling or shocks. Non-exercised mice were housed in cages without a cage wheel. Daily time and distance run, as well as total distance run for each mouse was collected and used to calculate average daily running time and distance, as well as average running speeds. Between 3 and 10 biological replicates were tested for each experimental group according to the amount of mice available since occasionally, data was unable to be collected for some adult mice as they would periodically chew through the speedometer wires. While biological replicates are low for a couple data sets, these did not have an impact on the overall results of the study. Data are presented as mean values ± S.E. Unpaired ttests were performed using SigmaStatTM v.3.5 (Systat Software Inc.) in order to determine statistically signifycant strain differences as well significance over time. Statistical significance was judged as p ≤ 0.05. Exercised and non-exercised mice were euthanized by cervical dislocation under inhaled anesthesia immediately after the end of the specific exercise period. Body mass and heart mass was quantified to determine any anatomical adaptations as a result of exercise.
2.2. Gene Expression
Gross muscle samples were taken from the left hind limb of each mouse to be used for gene expression studies. Total RNA was isolated from the muscle samples using a RNeasy Fibrous Tissue Mini kit (Qiagen). The RNA was converted to cDNA with ABI High Capacity cDNA kit according to the manufacturer’s protocols (Applied Biosystems). Expression levels of three genes were quantified: glucose transporter 4 (GLUT4), pyruvate dehydrogenase (PDHA1), and phosphofructokinase (PFKM). The primer sets used were Taqman Gene Expression Assays, Mm00436615_m1, Mm00468675_m1, and Mm00445461_m1, respectively (Applied Biosystems). Each primer set was carefully selected so that amplicons would cross exon boundaries to prevent contamination by genomic DNA. Assays were performed using realtime PCR techniques in triplicate from a total of three biological replicates. In each reaction, 8 ng of cDNA was used. Relative expression was determined from experimentally derived Ct values normalized to an endogenous control, β-glucuronidase, Mm00446953_m1, which is equally expressed in all samples. A relative standard curve analysis was used to correct Ct values, accounting for small variations in primer efficiency. Standard curve samples were made by pooling cDNA from one biological replicate of each experimental group and assaying concentrations ranging from 0.1 ng to 100 ng. Standard curves for all four primers are found in Figure S1. One-way analysis of variance (ANVOA) was used to assess the effects of exercise on gene expression within each exercised experimental set (1 week, 2 week, etc.); unpaired t-tests were used to determine significant changes in gene expression due to strain and age differences. Data are presented as mean values ± S.E. Statistical significance was judged as p ≤ 0.05. All statistics were determined using SigmaStatTM software.
3. RESULTS
To determine whether I/LnJ mice demonstrate exercise impairment as a result of the phka1 mutation, mice were allowed to run voluntarily for 1, 2, or 5 weeks. As shown in Figure 1, after only a two week exposure to the wheels, the cumulative time and distance run by the juvenile I/LnJ mice was significantly less than that of agematched wild type C57Bl6 mice. Moreover, after five weeks of exercise, the I/LnJ mice ran approximately one-half the cumulative time and distance of the wildtype mice, 116 ± 6 hours and 211 ± 23 km versus 194 ± 3 hours and 418 ± 4 km, respectively. The only significant results between the two strains of adult mice was that of running time after two weeks; I/LnJ mice ran 268 ± 64 min/day compared to 375 ± 15 min/day for wild-type (Figures 2(a) and (b)). However, while not statistically
 (a)
(a) (b)
(b)
Figure 1. Juvenile I/LnJ mice demonstrate a reduction in voluntary exercise over a 5 week training period. Cumulative running time (a) and distance (b) of I/LnJ mice (white bars) is compared to age-matched C57Bl6 mice (dark bars). Data are mean values ± S.E.; n = 2 - 10; *represents a significant difference when compared to age-matched mice at same training interval; p ≤ 0.05.
 (a)
(a) (b)
(b) (c)
(c)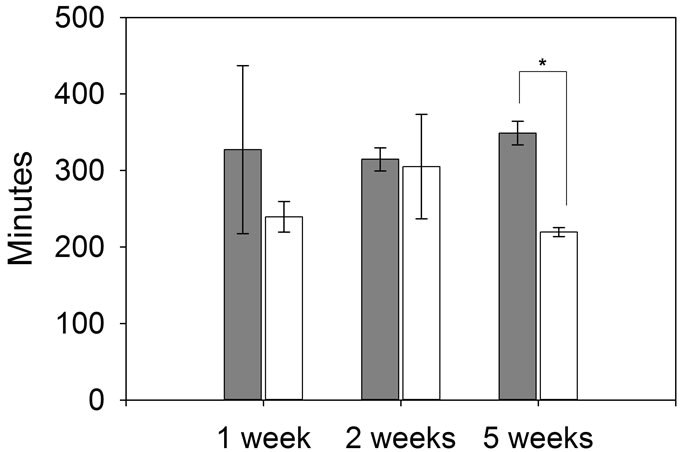 (d)
(d)
Figure 2. I/LnJ mice are unable to adapt to aerobic training. The average daily activity of adult I/LnJ mice (white hatched bars) is compared to adult C57Bl6 mice (dark hatched bars) in panels (a) and (b). Daily juvenile I/LnJ activity (white solid bars) is compared to juvenile C57Bl6 activity (dark solid bars) in panels (c) and (d). Data represents the mean distance run in kilometers ((a), (c)) and mean time run in minutes ((b), (d)) ± S.E.; n = 2 - 10; *represents a significant difference when compared to age-matched mice at same training interval; p ≤ 0.05. #represents a significant difference when compared to 1 week of same strain; p ≤ 0.05.
significant, it is clear that overall, adult I/LnJ mice run less time and distance on average per day across the entire training period than age-matched wild type mice. Juvenile I/LnJ mice also run less daily time and distance than controls. After five weeks of exercise, the juvenile I/LnJ mice ran 219 ± 6 min/day as compared to 349 ± 15 min/day for the wild-type and an average of 7 ± 1 vs 15 ± 2 km/day, respectively (Figures 2(c) and (d)). These data demonstrate that I/LnJ mice, both juvenile and adult, display significant exercise intolerance and do not adapt to aerobic training periods as compared to wild-type mice. It should be noted that the daily running activity levels in this study are consistent with other exercise studies in mice. In particular, at C57Bl6 mice 8 - 10 weeks old reportedly run between 3.1 and 16.2 km/day and 144 - 426 min/day [28]. The adult wild-type mice in our study ran farther after a five week training interval (18.6 km/day), but otherwise the average times and distances run were comparable to previous reports (Figure 2).
Interestingly, both juvenile and adult I/LnJ mice ran at speeds that were not statistically different from agematched wild type counterparts (Figure 3). I/LnJ mice ran 75% - 80% of the average speed of C57Bl6 mice after five weeks acclimation to exercise; wild-type and I/LnJ juvenile mice ran 43 ± 4 m/min and 32 ± 3 m/min respectively (Figure 3(a)), whereas wild-type and I/LnJ adult mice ran 48 ± 5 m/min and 39 ± 5 m/min, respectively (Figure 3(b)). The observed C57Bl6 results in this study are also consistent with running speeds reported for this strain previously [28]. Overall, the results of the voluntary free-running experiments suggest that the mutation in phka1 does not significantly impair speed (the ability to run fast) in I/LnJ mice, but instead diminishes stamina (the ability to run fast at long distance) as a result of suppressed glycogenolytic activity.
It is well known that physiological cardiac hypertrophy is a key remodeling event that occurs in response to isotonic exercise [29] and references therein], and voluntary running programs have been shown to induce physiological adaptations [28]. In order to investigate whether physiological cardiac remodeling was occurring in response to exercise in I/LnJ mice, heart weight/body weight (HW/BW) ratios were recorded (Table 1). While voluntary wheel activity did induce a significant increase in HW/BW in C57Bl6 mice between 1 week and 5 weeks of exercise (Table 1), the I/LnJ mice did not undergo any physiological adaptation. The absence of any quantitative endurance exercise-induced cardiac hypertrophy in the I/LnJ mice is likely a result of their inability to increase run duration and distance throughout the training interval. This is not surprising as other mutant mice that have lower exercise capacities also present with lower hypertrophic responses [30].
 (a)
(a) (b)
(b)
Figure 3. A mutation in phka1 does not significantly diminish running speed in I/LnJ mice. The average running speed of adult I/LnJ mice (white hatched bars) is compared to agematched C57Bl6 mice (dark hatched bars) in panel (a). Juvenile running speed differences are given in panel (b); I/LnJ = white bars and C57Bl6 = dark bars. Data represents mean values ± S.E., n = 2 - 10, n = 2 - 10; *represents a significant difference when compared to age-matched mice at same training interval; p ≤ 0.05. #represents a significant difference when compared to 1 week of same strain; p ≤ 0.05.
Although the I/LnJ mice have reduced exercise capacity, they are indeed capable of achieving moderate amounts of exercise in the absence of the ability to breakdown stored glycogen. This led us to hypothesize that the expression of key enzymes necessary for glucose transport and glycolytic flux may be upregulated in the I/LnJ mouse yielding a compensatory metabolic strategy for the chronic absence of muscle PhK. To test this hypothesis, we quantified expression of scla1, the gene that encodes glucose transporter 4 (GLUT4), the enzyme involved in glucose transport from the blood, pfkm, which encodes phosphofructokinase (PFK), the ratelimiting enzyme in glycolysis, and pdha1, which encodes pyruvate dehydrogenase (PDH), the enzyme involved in the conversion of pyruvate to acetyl CoA. Based upon

Table 1. Exercise does not induce physiological cardiac hypertrophy in juvenile I/LnJ mice. Data represents the mean value ± S.E.; n = 3 - 10.
RT-PCR analysis, there was no significant difference in the amount of gene expression for any of the three genes examined when wild-type C57Bl6 and I/LnJ mice were compared at the same training intervals (Figure 4). Additionally, when the transcriptional levels for each gene were evaluated over the course of the entire training interval, no changes were found within strains as a result of exercise (Figure 4). A distinct developmental difference in the expression of pdha between adult and juvenile mice, regardless of strain, was observed (Figure 4(c)). This is the first report, to our knowledge, that has identified a developmental regulation of PDHa expression, and is consistent with other studies reporting low levels of cardiac PDHa activity in neonatal rats as compared to adults [31].
4. CONCLUSION
The present study establishes that the I/LnJ mouse strain exhibits mild exercise intolerance with no compensatory increase in glycolytic gene expression when compared to healthy wild type mice and also confirms the hypothesis, from human studies, that a deficiency in phka1 does indeed result in a mild metabolic myopathy.
5. DISCUSSION
Recently, it has been suggested that the rare glycolgenolytic disorder GSD Type IX, which results from mutations in the human PHKA1 gene, should be clinically defined as a metabolic variant rather than a pathological myopathy [9,32,33]. (Note that in references 9 and 33, the disorder is classified as GSD Type VIII, an earlier classification for PhK deficiencies that is no longer used). By using the I/LnJ mouse as a model for GSD Type IX, our work also supports this conclusion. When mutant and wild-type mice were allowed to exercise voluntarily for either 1, 2 or 5 weeks, the most striking difference between the two strains was in the total time and total distance run; the I/LnJ mice could achieve only ~50% of the total distance that the wildtype mice ran after a 5 week training period. While the daily distances and time periods run by the two strains were initially similar, their activity levels diverged significantly over the five-week period. Ultimately, the wild type mice adapted to the training period, increasing their aerobic potential as expected, whereas I/LnJ mice could not. This data is consistent with the human form of GSD Type IX in which all patients present with mild to moderate exercise intolerance [9,33-35].
The lack of severity in the myopathy is noteworthy given that the I/LnJ mice only have 0.3% of the normal PhK activity and thus, cannot phosphorylate Pb; indeed previous work has shown that in response to epinephrine or exercise, no sufficient amounts of Pa are detectable in the muscle of I/LnJ mice although glycogenolysis does occur [36]. In fact, I/LnJ muscle degrades glycogen via allosteric nucleoside activation of Pb. In an elegant study by Rahim, et al., it was shown that tetanic stimulation of I/LnJ muscle results in a 60 - 100 fold increase in the nucleoside monophosphate, IMP, and that non-covalent IMP-mediated glycogenolysis takes place after forced swimming exercise [18,36]. Thus, it is highly likely that in the present study, the I/LnJ mice are capable of sustaining short periods of sub-maximal exercise due to allosteric activation of glycogenolysis, however outputs near that of wild type mice are impossible due to the inability to activate Pb via PhK phosphorylation, which yields higher and earlier peak rates of glycogenolysis [19]. Moreover, exercise studies undertaken with the muscle glycogen synthase knock out (MGSKO) mouse suggest that hepatic glycogen stores may be quantitatively more important in rodent exercise than skeletal muscle glycogen [37]. Like the MGSKO mice, the I/LnJ mice also show blunted lactate levels following exercise [19,37], thus we cannot rule out supplementary utilization of hepatic glycogen stores by the I/LnJ mouse. Regardless, the present study does demonstrate that muscle glycogenolysis is not inconsequential, and a deficiency in phka1 does yield a mild myopathy.
In exercising muscle, glucose equivalents provided by muscle glycogen can also be replaced by either oxidation of fatty acid or an increase in blood glucose uptake. The observation that lactate levels are blunted in I/LnJ skeletal muscle during exercise does suggest that other catabolic pathways may be active. In this study, we examined the expression of three key genes that mediate various rate limiting steps in glycolysis: GLUT4, the primary transporter for blood glucose uptake, PFK, the
 (a)
(a) (b)
(b)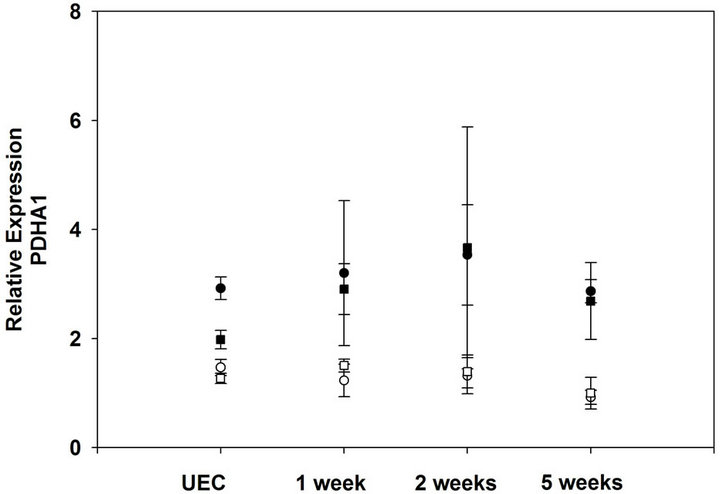 (c)
(c)
Figure 4. Changes in glycolytic gene expression does not compensate for the absence of muscle PhK in I/LnJ mice. Transcriptional levels of the following genes were quantified from I/LnJ (squares) or C57Bl6 (circles) skeletal muscle following the indicated exercise period: scla1 (Panel (a)), pfkm (Panel (b)), and pdha1 (Panel (c)). Relative expression is calculated using a relative standard curve (see Methods) and represents the mean of three biological replicates ± S.E. UEC = unexercised control. No statistically significant differences in expression were identified.
key rate-limiting enzyme in glycolysis, and PDHa, a subunit involved in the conversion of pyruvate to acetyl CoA. When quantified, there was no change in transcript levels both between the wild type and the I/LnJ mice for any exercise interval, and also within each strain over the duration of the training period. These results indicate that the I/LnJ mice do not compensate the observed deficit in glycogenolysis by an increase in glycolytic enzyme transcription. The fact that no change was observed in GLUT4 expression is surprising given that it has been shown to be up regulated at both the mRNA and protein level in response to exercise [23]; this observed discrepancy is most likely due to a difference in the exercise stimulus between the two studies. In the study by Ren, et al. rats swam non-voluntarily for 3 hours at a time for two days prior to quantification of GLUT4. Moreover, the increase in GLUT4 transcription in response to exercise has been shown to occur following running or swimming exercise programs 6 to 12 weeks long. Therefore, it is possible that the time frame of our study simply was not long enough to result in a genetic adaptation [24,25].
6. ACKNOWLEDGEMENTS
The authors would like to thank Dr. David Allen for critical readings of the manuscript. Funding from grant P20GM103436-12 from the NIGMS of the NIH is gratefully acknowledged.
REFERENCES
- Pickett-Gies, C.R. and Walsh, D.A. (1986) Phosphorylase kinase. In: Boyer, P.O. and Krebs, E.G., Eds., The Enzymes, Academic Press, Orlando, 395-459.
- Krebs, E.G. (1993) Nobel lecture. Protein phosphorylation and cellular regulation I. Bioscience Reports, 13, 127-142. doi:10.1007/BF01149958
- Brushia, R.J. and Walsh, D.A. (1999) Phosphorylase kinase: The complexity of its regulation is reflected in the complexity of its structure. Frontiers in Bioscience, 4, D618-D641. doi:10.2741/Brushia
- Heilmeyer Jr., L.M. (1991) Molecular basis of signal integration in phosphorylase kinase. Biochimica et Biophysica Acta, 1094, 168-174. doi:10.1016/0167-4889(91)90005-I
- Rice, N.A. and Carlson, G.M. (2001) Phosphorylase kinase. Wiley Encyclopedia of Molecular Medicine, John Wiley and Sons, Inc., New York, 2487-2490.
- Wehner, M., Clemens, P.R., Engel, A.G. and Kilimann, M.W. (1994) Human muscle glycogenosis due to phosphorylase kinase deficiency associated with a nonsense mutation in the muscle isoform of the alpha subunit. Human Molecular Genetics, 3, 1983-1987. doi:10.1093/hmg/3.11.1983
- Wuyts, W., Reyniers, E., Ceuterick, C., Storm, K., de Barsy, T. and Martin, J.J. (2005) Myopathy and phosphorylase kinase deficiency caused by a mutation in the PHKA1 gene. American Journal of Medical Genetics Part A, 133, 82-84. doi:10.1002/ajmg.a.30517
- Burwinkel, B., Hu, B., Schroers, A., Clemens, P.R., Moses, S.W., Shin, Y.S., Pongratz, D., Vorgerd, M. and Kilimann, M.W. (2003) Muscle glycogenosis with low phosphorylase kinase activity: Mutations in PHKA1, PHKG1 or six other candidate genes explain only a minority of cases. European Journal of Human Genetics, 11, 516-526. doi:10.1038/sj.ejhg.5200996
- Orngreen, M.C., Schelhaas, H.J., Jeppesen, T.D., Akman, H.O., Wevers, R.A., Andersen, S.T., Ter Laak, H.J., van Diggelen, O.P., DiMauro, S. and Vissing, J. (2008) Is muscle glycogenolysis impaired in X-linked phosphorylase b kinase deficiency? Neurology, 70, 1876-1882. doi:10.1212/01.wnl.0000289190.66955.67
- Abarbanel, J.M., Bashan, N., Potashnik, R., Osimani, A., Moses, S.W. and Herishanu, Y. (1986) Adult muscle phosphorylase “b” kinase deficiency. Neurology, 36, 560-562. doi:10.1212/WNL.36.4.560
- Cohen, P.T., Burchell, A. and Cohen, P. (1976) The molecular basis of skeletal muscle phosphorylase kinase deficiency. European Journal of Biochemistry, 66, 347-356. doi:10.1111/j.1432-1033.1976.tb10524.x
- Le Marchand-Brustel, Y., Cohen, P.T. and Cohen, P. (1979) Insulin activates glycogen synthase in phosphorylase kinase deficient mice. FEBS Letters, 105, 235-238. doi:10.1016/0014-5793(79)80619-5
- Bender, P.K. and Lalley, P.A. (1989) I/Lyn mouse phosphorylase kinase deficiency: Mutation disrupts expression of the alpha/alpha’-subunit mRNAs. Proceedings of the National Academy of Sciences of the USA, 86, 9996- 10000. doi:10.1073/pnas.86.24.9996
- Bender, P.K. (1991) Phosphorylase kinase activity in I/strain neonatal skeletal muscle with a deficiency in alpha/alpha’ subunit mRNAs. Biochemical and Biophysical Research Communications, 179, 707-712. doi:10.1016/0006-291X(91)91430-K
- Schneider, A., Davidson, J.J., Wullrich, A. and Kilimann, M.W. (1993) Phosphorylase kinase deficiency in I-strain mice is associated with a frameshift mutation in the alpha subunit muscle isoform. Nature Genetics, 5, 381-385. doi:10.1038/ng1293-381
- Lyon Jr., J.B., Porter, J. and Robertson, M. (1967) Phosphorylase b kinase inheritance in mice. Science, 155, 1550-1551. doi:10.1126/science.155.3769.1550
- Gross, S.R. and Mayer, S.E. (1974) Characterization of the phosphorylase b to a converting activity in skeletal muscle extracts of mice with the phosphorylase b kinase deficiency mutation. The Journal of Biological Chemistry, 249, 6710-6718.
- Rahim, Z.H., Perrett, D., Lutaya, G. and Griffiths, J.R. (1980) Metabolic adaptation in phosphorylase kinase deficiency. Changes in metabolite concentrations during tetanic stimulation of mouse leg muscles. Biochemical Journal, 186, 331-341.
- Danforth, W.H. and Lyon Jr., J.B. (1964) Glycogenolysis during tetanic contraction of isolated mouse muscles in the presence and absence of phosphorylase A. The Journal of Biological Chemistry, 239, 4047-4050.
- Charron, M.J., Brosius 3rd, F.C., Alper, S.L. and Lodish, H.F. (1989) A glucose transport protein expressed predominately in insulin-responsive tissues. Proceedings of the National Academy of Sciences of the USA, 86, 2535- 2539. doi:10.1073/pnas.86.8.2535
- James, D.E., Strube, M. and Mueckler, M. (1989) Molecular cloning and characterization of an insulin-regulatable glucose transporter. Nature, 338, 83-87. doi:10.1038/338083a0
- Fushiki, T., Wells, J.A., Tapscott, E.B. and Dohm, G.L. (1989) Changes in glucose transporters in muscle in response to exercise. American Journal of Physiology, 256, E580-E587.
- Ren, J.M., Semenkovich, C.F., Gulve, E.A., Gao, J. and Holloszy, J.O. (1994) Exercise induces rapid increases in GLUT4 expression, glucose transport capacity, and insulin-stimulated glycogen storage in muscle. The Journal of Biological Chemistry, 269, 14396-14401.
- Ploug, T., Stallknecht, B.M., Pedersen, O., Kahn, B.B., Ohkuwa, T., Vinten, J. and Galbo, H. (1990) Effect of endurance training on glucose transport capacity and glucose transporter expression in rat skeletal muscle. American Journal of Physiology, 259, E778-E786.
- Rodnick, K.J., Henriksen, E.J., James, D.E. and Holloszy, J.O. (1992) Exercise training, glucose transporters, and glucose transport in rat skeletal muscles. American Journal of Physiology, 262, C9-C14.
- Rovira, J., Irimia, J.M., Guerrero, M., Cadefau, J.A. and Cusso, R. (2012) Upregulation of heart PFK-2/FBPase-2 isozyme in skeletal muscle after persistent contraction. Pflügers Archiv—European Journal of Physiology, 463, 603-613. doi:10.1007/s00424-011-1068-5
- Zonderland, M.L., Bar, P.R., Reijneveld, J.C., Spruijt, B.M., Keizer, H.A. and Glatz, J.F. (1999) Different metabolic adaptation of heart and skeletal muscles to moderate-intensity treadmill training in the rat. European Journal of Applied Physiology and Occupational Physiology, 79, 391-396. doi:10.1007/s004210050527
- Allen, D.L., Harrison, B.C., Maass, A., Bell, M.L., Byrnes, W.C. and Leinwand, L.A. (2001) Cardiac and skeletal muscle adaptations to voluntary wheel running in the mouse. Journal of Applied Physiology, 90, 1900-1908.
- McMullen, J.R. and Jennings, G.L. (2007) Differences between pathological and physiological cardiac hypertrophy: Novel therapeutic strategies to treat heart failure. Clinical and Experimental Pharmacology and Physiology, 34, 255-262. doi:10.1111/j.1440-1681.2007.04585.x
- Horckmans, M., Leon-Gomez, E., Robaye, B., Balligand, J.L., Boeynaems, J.M., Dessy, C. and Communi, D. (2012) Gene deletion of P2Y4 receptor lowers exercise capacity and reduces myocardial hypertrophy with swimming exercise. American Journal of Physiology—Heart and Circulatory Physiology, 303, H835-H843. doi:10.1152/ajpheart.00256.2012
- Sugden, M.C., Langdown, M.L., Harris, R.A. and Holness, M.J. (2000) Expression and regulation of pyruvate dehydrogenase kinase isoforms in the developing rat heart and in adulthood: Role of thyroid hormone status and lipid supply. Biochemical Journal, 352, 731-738. doi:10.1042/0264-6021:3520731
- Haller, R.G. (2008) Fueling around with glycogen: The implications of muscle phosphorylase b kinase deficiency. Neurology, 70, 1872-1873. doi:10.1212/01.wnl.0000312284.43608.46
- Echaniz-Laguna, A., Akman, H.O., Mohr, M., Tranchant, C., Talmant-Verbist, V., Rolland, M.O. and Dimauro, S. (2010) Muscle phosphorylase b kinase deficiency revisited. Neuromuscular Disorders, 20, 125-127. doi:10.1016/j.nmd.2009.11.004
- Chen, S.T., Chen, H.L., Ni, Y.H., Chien, Y.H., Jeng, Y.M., Chang, M.H. and Hwu, W.L. (2009) X-linked liver glycogenosis in a Taiwanese family: Transmission from undiagnosed males. Pediatrics & Neonatology, 50, 230-233. doi:10.1016/S1875-9572(09)60068-1
- Pears, J.S., Jung, R.T., Hopwood, D., Waddell, I.D. and Burchell, A. (1992) Glycogen storage disease diagnosed in adults. The Quarterly Journal of Medicine, 82, 207-222.
- Lyon Jr., J.B. and Porter, J. (1963) The relation of phosphorylase to glycogenolysis in skeletal muscle and heart of mice. The Journal of Biological Chemistry, 238, 1-11.
- Pederson, B.A., Cope, C.R., Schroeder, J.M., Smith, M.W., Irimia, J.M., Thurberg, B.L., De Paoli-Roach, A.A. and Roach, P.J. (2005) Exercise capacity of mice genetically lacking muscle glycogen synthase: In mice, muscle glycogen is not essential for exercise. The Journal of Biological Chemistry, 280, 17260-17265. doi:10.1074/jbc.M410448200
SUPPLEMENTARY

NOTES
*Corresponding author.