Advances in Biological Chemistry
Vol. 2 No. 2 (2012) , Article ID: 18974 , 14 pages DOI:10.4236/abc.2012.22016
Phosphatidylglycerol-containing ER-transport vesicles built and restore outer mitochondrial membrane and deliver nuclear DNA translation products to generate cardiolipin in the inner mitochondrial membrane
![]()
Research Center, University of Medicine and Dentistry of New Jersey, Newark, USA
Email: slomiaam@umdnj.edu
Received 8 February 2012; revised 2 March 2012; accepted 12 March 2012
Keywords: ER-transport vesicles; mitochondrial membranes biogenesis; transport of nuclear DNA translation products; cardiolipin synthase; cell organelles repair
ABSTRACT
Phosphatidylglycerol (PG) an important membrane phospholipid required for the synthesis of diphosphatidylglycerol (DPG) commonly known as cardiolipin (CL) was identified in the fraction of endoplasmic reticulum (ER)-derived transport vesicles which had no affinity for Golgi. The vesicles were produced in the presence of Brefeldin A (BFA), the agent known to inhibit ER-Golgi transport, and found to display affinity to mitochondria. The analysis revealed that their cargo was not containing proteins that are transported to Golgi, and that their membrane was free of phosphatidylinositol (PI) and ceramides (Cer). The incubation of PG-containing transport vesicles with mitochondria afforded incorporation of their membrane into the Outer Mitochondrial Membrane (OMM) and formation of lysophosphatidylglycerol (LPG). In turn, upon further incubation with fresh transport active cytosol, the mitochondrial LPG was converted to PG. The results of analysis of the OMM, Inner Mitochondrial Membrane (IMM) and Inner Mitochondrial Space Components (IMSC) strongly suggest that PG-containing transport vesicles deliver nuclear DNA translation products to the IMSC and thus facilitate CL synthesis in the IMM. In summary, our studies provide evidence that ER-generated PG-enriched transport vesicles represent the general pathway for restitution of mitochondrial membranes and the delivery of nuclear DNA translation products that generate CL, and thus sustain the mitochondrial matrix CL-dependent metabolic reactions.
1. INTRODUCTION
Endoplasmic Reticulum (ER) is the largest cell organelle that in secretory epithelia represents close to 50% of total membranes [1]. The expansive size of ER membrane reflects the manifold functions associated with this organelle, including synthesis of the faithfully recreated biomembranes that restore cell organelles and cell’s membrane, and with uncompromised fidelity recondition the cell-specific physiological functions. To accommodate these diverse organelle-specific assignments, the ER organelle continuum engages its translation domains in the assembly of the cargo and the vesicular membranes destined to the specific intracellular or the cell membrane sites [2,3]. The rule that guides these processes is in the decoding of nuclear signals that are materialized in the discharge of the specific mRNAs, their translation and the cotranslational intercalation of the integral membrane proteins within concomitantly synthesized organellespecific membrane lipids [4]. Such explicit and highly controlled membrane’s assembly satisfies fidelity of cell structure restitution, and retention of the celland organelle-characteristic features. However and because, the mRNA translation processes are continually tackled as an independent from ER membrane scaffold assemblies, the orchestrated series of events that produce signal-specific products are still not visualized in their entirety as a single and unified complex event. Moreover, at the outset of the cellular transport investigations, an idea prevailed that featured several vesicular surface proteins as the only necessary markers of the specific transporters [5]. This further constrained the view on cellular transport specificity and hampered the research on transport vesicles complexity [6]. Hence, many attributes of the transport initiation, transport vesicles composition, and the critical role of the cell cytosol in the features of generated cell membranes and their cargo remained unrecognized.
In the concept developed from our findings, the range of events that determine cell-specific transporters is defined by concomitant nuclear signal-induced translation of the cargo protein and the membrane-intercalated protein into the concurrently synthesized membrane lipids [2-4,7]. The critical breakthrough in our investigation occurred with realization that for the preservation of cellular and organellar identity, the nuclear signals initiate in ER the production of a numerous non-identical transport vesicles which fulfill the tasks of cell repair, restitution, and secretory functions [3]. Following this thought, we identified transport vesicles destined for apical, basolateral and endosomal delivery; all were produced by ER and initially displayed affinity for Golgi, but upon their membrane maturation in Golgi became destination-specific [3,8-11]. However, neither in our earlier study nor in the investigations by others, the path to restore mitochondria as vesicles-dependent event was grasped [10- 13]. Up till now, the ER-mitochondria transport was pictured as a series of the ER-mitochondria close contacts that supposedly maintain transfer of mitochondria-essential calcium, lipids and proteins [14,15]. While such contacts between organelles could accommodate transfer of calcium, the process of mitochondrial membranes repair and restitution, and the demand to coordinate nuclear gene expression and maintain regulatory processes in response to cellular need for ATP synthesis were not plausible [3,4,16-18]. The mitochondrial oxidative phosphorylation system is dependent on both nuclear and mitochondrial DNA (mtDNA). While mammalian mtDNA encodes 13 proteins that all are subunits of the oxidative phosphorylation system, the rest of mitochondrial proteome depends on nuclear DNA products [12,13,15]. Therefore, highly coordinated delivery system such as ER vesicles-controlled delivery must operate to provide nuclear products in the required time and quantity [15]. Since such carefully measured and regulated responses of ER-Golgi pathway handling multiplicity of the specific transport vesicles was identified, we concentrated on the potential involvement of the ER vesicles in the delivery of nuclear DNA products and membranes to mitochondria. Toward that end, we analyzed the fraction of ER-assembled vesicles remaining in the cytosol after transport to Golgi was accomplished, and on the vesicles produced when BFA inhibited the Golgi pathway [19-21]. The analyses revealed that the vesicles remaining in cytosol, after completion of ER-Golgi transport or in BFAinhibited transport, consisted of lipids enriched with phosphatidylglycerol (PG), while phosphatidylinositol (PI) and ceramides (Cer) were not detectible. Moreover, these vesicles were not displaying affinity for ER or the organelles which were renewable by ER to Golgi-channeled transport described earlier [2,9,10]. Instead, they displayed the affinity to mitochondria and fused with mitochondrial membrane.
In our interpretation, these results revealed that mitochondrial membranes renewal is accomplished through the delivery of ER-assembled mitochondria-specific transport vesicles. Collectively, the investigation supports our initial concept regarding universal mechanism for cell repair, renewal and restitution. The process is highly specific, dictated by the nuclear release of the signals materialized in the synthesis of multiplicity of the transport vesicles, which maintain organellar repair and retain uncompromised compositional identity of the membrane that is designed for the specific cell, its organelles, and its function.
2. MATERIALS AND METHODS
2.1. Preparation of Cells for Subcellular Fractionation
For the isolation of cell components, the initial step of fragmentation of the structure is crucial and a method is required that gives minimum breakage. Hence, in the method described herein for the isolation of Outer Mitochondrial Membrane (OMM) and Inner Mitochondrial Membrane (IMM) we used a technique which allowed us to purify cell organelles with minimum fragmentation of their structure so that Cell Cytosol (CC) was not admixed with organellar components and that cell membranes were not admixed with intracellular membranes. Moreover, in preparation of mitochondria we aimed to isolate undamaged structure in order to detach OMM as single sheet, unbroken inner membrane (IMM), and recover Inner Mitochondrial Space Content (IMSC), to determine its activity toward synthesis, deacylation and reacylation of membrane lipids.
The cells were prepared from rat gastric mucosa and the liver as described previously [2,7,9,10]. The single cells that were separated from larger debris with aid of specific cell size nylon mesh were centrifuged at 50 × g for 2min, washed twice with the enzyme-free medium, twice with the Minimum Essential Medium (MEM) and counted in hemocytometer. Thus prepared cells were then incubated in MEM for 3 hours with or without radiolabel and used for preparation of nuclei [7], subcellular organelles, cell cytosol [2,7,9,11] and cellular membranes [3,11]. In the experiments dedicated to the determination of lipid synthesis with cell cytosol derived from gastric epithelial cells, hepatocytes, or RNase treated cytosol, the preparations of nuclei, mitochondria, ER, Golgi or other organelles and cell membranes were additionally rinsed with phosphate buffered saline (PBS) and ureaPBS in order to remove the associated residual cytosolic proteins that otherwise would remain on the membranes. Thus prepared subcellular organelles and membranes were used for experiments on transport vesicles synthesis [3,11], and the preparation of OMM and IMM [16]. The synthesis of phospholipids and protein was determined using radiolabeled [3H]inositol, [3H] arachidonate, [3H]choline, [3H]serine, [3H]palmitate and [32P]ATP [2,4,7,9-11]. The radiolabeled ER transport vesicles fusion with Golgi, ER, mitochondria were performed in medium containing cold cell cytosol (CC) at concentration of 15 mg protein/ml of incubation mixture enriched with 50 μM ATP, 250 μM CTP, 50 μM GTP, 5 mM creatine phosphate, 8.0 IU/ml creatine kinase, and where indicated 25 μg/ml RNase, 10 μM UDP-Glc and 10 μM palmitoyl CoA [2,4,7,9-11].
2.2. Preparation of Transport-Active Cell Cytosol (CC)
The viable cells, homogenized for 10 sec at 600 rpm in 3 volumes of buffer containing 0.25 M sucrose; 50 mM TRIS-HCl (pH 7.4), 25 mM magnesium acetate and 10 mM each of aprotinin, leupeptin, chemostatin, and 1 mM phenylmethylsulfonylfluoride, were centrifuged at 5000 × g for 15 min. The supernatant, diluted with 2 volumes of homogenization buffer, was re-centrifuged at 10,000 × g for 20 min. The resulting supernatant was then subjected to centrifugation at 100,000 × g for 1 h. Thus obtained soluble fraction was adjusted to 15 - 18 mg protein/ml, admixed with an ATP generating system consisting of 40 mM ATP, 200 mM creatine phosphate, 2000 units/ml creatine phosphokinase, and referred to as transport active cell cytosol or active cytosol (CC).
2.3. Preparation of Cellular Organelles and Membranes
The cell membranes and subcellular organelles (mitochondria, ER, Golgi) were recovered from the cold or radiolabeled cells as described earlier [2,4,7,9-11]. The ER and Golgi organelles sediment, remaining after separation of nuclei, mitochondria and cell cytosol, was suspended in buffer containing 0.2 M PIPES (pH 6.9), 2 M glycerol, 1 mM EGTA and 1 mM magnesium acetate and applied on the top of discontinuous gradient of 2.0/1.5/ 1.3/1.0 M sucrose and centrifuged at 100,000 × g for 16 h. The cell membranes were recovered from 1.0M sucrose, Smooth Endoplasmic Reticulum (SER) from 1.3 M sucrose, RER from 1.5 M sucrose and Golgi from the top of the 2.0 M sucrose. Each sucrose-separated fraction was subjected to further purification. The cell membranes were washed with original PIPES buffer and centrifuged at 3000 rpm for 2 min. To separate apical epithetlial membranes, the buffer was adjusted with 0.2% Triton X-100 and the mixture incubated at 4˚C for 5 min [4,11]. This treatment resulted in breaking up the phospholipids-rich membranes into smaller segments and that allowed us to separate apical membranes containing cholesterol, glycosphingolipids and glycoproteins. The latter membranes were recovered by low speed centrifugation at 3000 rpm for 2 min.
2.4. Generation and Purification of Transport Vesicles
ERand Golgi-derived transport vesicles were generated in the presence of radiolabeled precursors according to procedure described previously [2,4,7,9-11], and where indicated, in the presence of Brefeldin A (BFA) [20]. The ER or Golgi membranes mixed with cytosol, ATP-generating system, UTP, CTP GTP, fatty acyl CoA and water soluble cold or radiolabeled lipids precursors, were incubated for 30 min at 37˚C, centrifuged over 0.3 M sucrose and treated with stripping buffer at 2˚C for 15 min followed by centrifugation at 10,000 × g for 10 min to separate transport vesicles from ER or Golgi membranes. The separated from maternal membranes transport vesicles were recovered from the supernatant resulting from centrifugation of the supernatant mixture at 150,000 × g for 60 min. The crude fraction of the transport vesicles was suspended in 55% sucrose, overlaid with 55% - 30% gradient and centrifuged at 150,000 × g for 16 h. The purified transport vesicles were recovered from the gradients as reported earlier [2,4,7,9-11].
2.5. Fusion of Transport Vesicles with Golgi, Mitochondria and ER
One volume of radiolabeled ER transport vesicles (1.3 - 1.5 mg protein/ml) was suspended in one volume of active cytosol (15 mg protein/ml), and added to one volume of cold cell organelles (5 mg protein/ml). The reaction was allowed to proceed from 0 - 30 min at 4˚C (control) and at 37˚C in the presence of ATP regenerating system consisting of 40 mM ATP, 200 mM creatine phosphate, 2000 units/ml of creatine phosphokinase, or in the ATP depleting system containing 5 mM glucose and 500 units/ml hexokinase. After incubation, the respective organelles were recovered by centrifugation through three volumes of 0.5 M sucrose at 3000 rpm for 5 min. The vesicles recovered from the supernatant after incubation with Golgi were purified on 55% - 30% sucrose gradient and used in fusion experiments with cold mitochondria. One volume of the recovered vesicles (0.9 - 1.1 mg/ml) was resuspended in one volume of CC (15 mg/ml) and added to one volume of purified mitochondria (5 mg/ml) and the reaction was allowed to proceed for 30 min under same conditions as described for the ER vesicles fusion with Golgi. In the experiments estimating en bloc fusion of transport vesicles with Golgi and/or mitochondria, the associated but not fused vesicles were released from the membrane by subjecting the membrane fraction to treatment with 2 M urea at 4˚C and then the recovered organelles were centrifuged through 0.5 M sucrose, washed and subjected to radiolabeled lipid analysis.
2.6. Isolation and Purification of Mitochondrial Membranes
The mitochondria were isolated from the hepatocytes derived from five perfused rat livers subjected to subcellular fractionation that afforded preparation of nuclei, mitochondria, Golgi, ER and organelle-free cell cytosol as described in [2,4,7,9-11] and above. From the initial fraction the fluffy layer of broken mitochondria and ER microsomes that covered the crude mitochondrial pellet was suctioned off, the pellet was gently resuspended in five volumes of medium consisting of 0.07 M sucrose, 0.21 M mannitol, 0.1 mM disodium EDTA, and 1mM TRIS, pH 7.2. and the suspension was spun for 10 min at low speed (500 × g), the supernatant recovered and spun for 10 min at 9000 × g [16]. This manipulation was repeated three times. Thus purified preparation of mitochondria containing only occasional broken mitochondria, ER membranes, or lysosomes was used for breaking and detaching the Outer Mitochondrial Membrane (OMM). The mitochondria were swollen for 20 min at 4˚C in 20 ml of swelling medium (20 mM phosphate buffer, pH 7.2). The swelling was initiated gradually by suspending the pellet in very small amount of swelling medium and progressively, over 3 - 5 min, diluting the mixture to final volume. After 20 min of treatment, the preparation was spun for 20 min at 17,000 rpm (35,000 × g), the pellet and the supernatant were separately recovered for further purification. The supernatant containing soluble the Inner Mitochondrial Space Components (IMSC) was centrifuged at 100,000 × g, filtered and concentrated in similar manner as in preparation of CC. The pellet was resuspended in fresh swelling buffer containing 0.02% albumin, and the Inner Mitochondrial Membranes (IMM) were sedimented at 4000 rpm (1900 × g) for 15 min. The OMM contained in the supernatant were concentrated by spinning at 17,000 × g, the pellet resuspended in 4.0 ml of 20 mM phosphate buffer, pH 7.2 and subjected to discontinuous sucrose gradient fractionation.
2.7. Discontinuous Sucrose Gradient
A three layered gradient was formed in Beckman SW39 tubes by layering successively sucrose solutions prepared in 20 mM phosphate buffer pH 7.2 consisting of 1.2 ml of 51.3 g/100ml sucrose solution (density 1.142), 1.2 ml of 37.7 g/100ml sucrose (density 1.142) and 1.2 ml of 25.2 g/100ml sucrose (density 1.094). One ml of concentrated OMM was layered on the top of the gradient and centrifuged at 37,500 rpm (115,000 × g) for one hour.
This resulted in separation of the membranes into three bands. From the top to bottom, the bands consisted of OMM small fragments (A), large sheets of OMM (B) and IMM with fragments of OMM (C), respectively. The identification of the isolated fraction is based on the electron microscopic studies published earlier [16]. To remove sucrose, the separated fractions were diluted in 20 mM phosphate buffer, pH 7.2 and centrifuged at 17,000 rpm (35,000 × g) for one hour. Then the IMM fraction recovered at 4000 rpm and the A, B, and C fractions recovered from discontinuous sucrose gradient were resuspended in 0.5 ml of the same buffer and used for enzyme assays and lipid analysis. The assays were performed in the presence of the IMSC or the CC components.
3. RESULTS
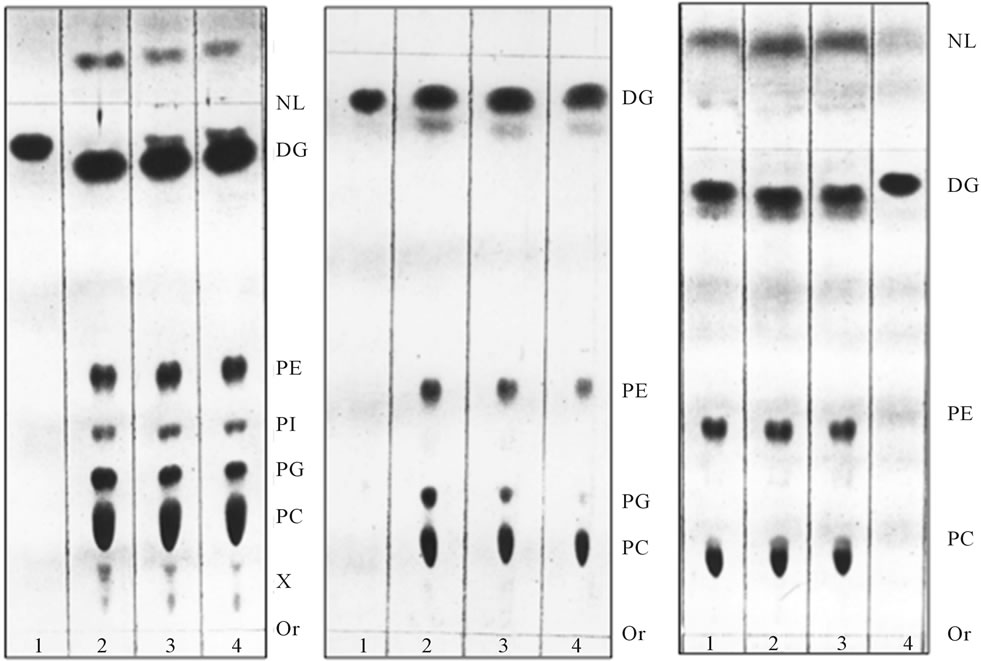
As demonstrated in earlier investigations of intracellular transport [20], the treatment with Brefeldin A (BFA) (5 - 10 mg/ml) afforded substantively different ER transport vesicles. Their protein composition was dissimilar with Golgi fusing vesicles, and their amount was reduced by up to 50%. The produced vesicles contained up to 70% less PI and Cer and their fusion with Golgi was reduced to 20% of the normal BFA-free control. The analysis of the vesicles that remained in the cytosol in BFA experiments and BFA-free controls, revealed even more pronounced differences reflected in their lipid composition. Namely, the vesicles recovered from the cytosol contained phosphatidylglycerol (PG), whereas those containing phosphatidylinositol (PI) and ceramides (Cer) were absent when ER-produced vesicular mixtures were first subjected to fusion with Golgi (Figure 1).
The PG-containing vesicles from BFA experiments and BFA-free controls recovered form cytosol after fusion reaction with Golgi were not reacting with ER or the organelles replenished by Golgi transport pathway, and the vesicles remained in the cytosol (Figure 2).
Since the consecutive attempts to fuse PG-enriched vesicles from BFA experiments or BFA-free controls with Golgi or ER were not successful, we concluded that the PG-containing vesicular fraction recovered from the cytosol does not represent retrograde transporters. In distinction, however, the PG-containing entities remaining in the cytosol reacted with mitochondria. Normally, as shown in Figure 3, the mitochondrial membranes contain substantial amount of PG, LPG and CL, and are practically devoid of PI and Cer.
Cardiolipin (CL), the prominent component of mitochondrial membrane, however, was not delivered or identified among the lipids of PG-containing transport vesicles that displayed affinity for mitochondrial membrane (Figures 1(b) and 2) or was identified in any other
 (a) (b) (c)
(a) (b) (c)
Figure 1. [3H] palmitate-labeled ER transport vesicles generated as described in Methods. Panel (a) illustrates lipid composition of the purified ER vesicles before fusion reaction with Golgi; Panel (b) illustrates lipid composition of the vesicles which remained in cytosol after reaction with Golgi; and Panel (c) illustrates radiolabeled lipids remaining in ER after formation of transport vesicles. In each panel, the lipids were extracted from three consecutive vesicular fractions recovered from 55% - 30% sucrose gradient (a, b) and ER (c). As shown earlier [2,4,7,9-11], the transport vesicles containing PI and Cer in their membrane fused with Golgi membranes, the vesicles containing PG (shown in (b)) remained in cytosol. Thin-layer chromatography was performed on 10 × 20 cm Silica H plates prescored and prewashed with solvent mixtures used for lipid separation and then activated at 85˚C overnight. The lipid extract from isolated fractions of transport vesicles (Panels (a) and (b)) and ER residue (panel (c)) were prepared in 10 μl of chloroform/methanol mixture (1/1 v/v) and the plate was developed up to 3/4 of length with solvent mixture of chloroform/methanol/water (65/35/5, v/v/v), dried and developed to within 0.5 cm of the plate top in hexane/diethyl ether/acetic acid (85/20/2, v/v/v). The iodine stained plates were subjected to radiolabel quantitation and to charring as describe previously [2,3,8-11]. The lipids were identified by parallel running of the phospholipid and neutral lipid standards. The lipid abbreviation, from the top, are: NL-neutral lipids, DG-diacylglycerides and ceramides, PE-phosphatidylethanolamine, PI-phosphatidy-linositol, PG-phosphatidylglycerol, PC-phosphatidylcholine, X-tentatively determined with ninhydrin spray as lysophosphatidylcholine (LPC) lower spot (ninhydrin negative), and lysophosphatidylserine (LPS) and/or lysophosphatidylethanolamine (LPE, ninhydrin positive), Or-origin.
cell organelles or cell membranes characterized previously [2-4,7,9-11].
The experiments tracing lipid conversions following PG transport vesicles fusion with mitochondrial membrane revealed that the process was accompanied by the synthesis of CL (Figure 4). Following incubation with CL-free radiolabeled PG transport vesicles, the radiolabeled CL was identified in the mitochondrial lipid profile. Moreover, the incorporation of PG transport vesicle membrane into the mitochondrial membrane was accompanied by a steady or even increased amount of lysophosphatidylglycerol (LPG) (Figure 4).
Interestingly, fusion reactions of the Golgi-specific ER transport vesicles and those delivered to plasma membrane, also produced lysophospholipids [11,22]. However, the former and latter produced lysophosphatidylcholine (LPC) and lysophosphatidylinositol phosphate (LPIP), respectively, whereas LPG presence was only observed in the mitochondrial membranes. Also, as evident from Figure 4, the fusion with mitochondria has not produced lysolipids of PA, PE or PC.
Considering the fact that the vesicles were radiolabeled with palmitate which in animal-derived phospholipids, primarily occupies sn-1 position of glycerol, it appeared that the lysolipids in mitochondrial membranes were generated by the action of phospholipase A2 (PLA2). Hence, a common cellular feature that determined fusion was revealed. The cytosolic phospholipid-specific PLA2
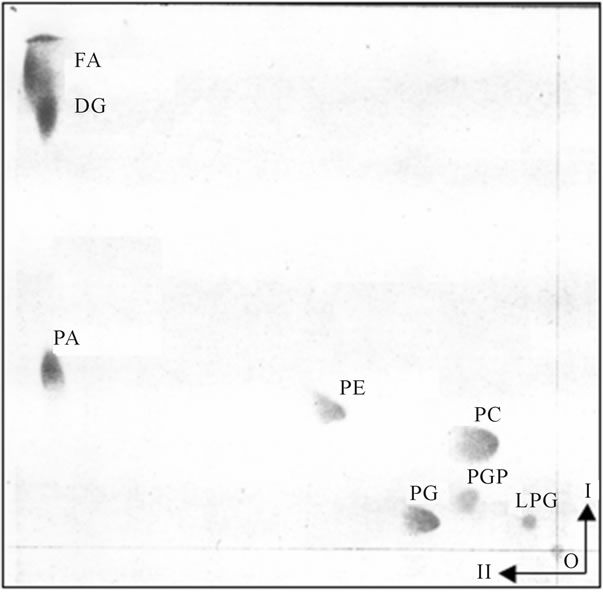
Figure 2. The PG-containing transport vesicles recovered from cytosol after fusion reaction with Golgi and ER. The two dimensional thin layer chromatography was performed on 10 × 10 cm high performance thin layer plates (HPTLC) prewashed with solvent mixtures used for lipid separation and activated as described for Figure 1. The lipid extracts from the purified transport vesicles that remained in the cytosol were applied to lower right corner of the plate and the plate was developed in I-solvent system consisting of chloroform/methanol/concentrated ammonium hydroxide (65/27/5, v/v/v). After drying in 37˚C oven for 1h, the plate was subjected to second (II) development in solvent system consisting of chloroform/acetone/ methanol/acetic acid/water (15/20/5/5/2.5, by vol.). The abbreviations placed on the plate correspond to following lipids: from the upper left corner FA-free fatty acids, DG-diacylglycerides, PA-phosphatidic acid, PE-phosphatidylethanolamine, PC-phosphatidylcholine, PG-phosphatidylglycerol, PGP-phosphatidylglycerol phosphate, LPG-lysophosphatidylglycerol. The lipids were identified using specific lipid standards and specific radiolabeled standards as described previously and under Figure 1.
appeared to govern fusion and its specificity with cellular organelles, and cell membranes [11,22]. The demonstration of the phospholipid-specific PLA2 activity also categorized fusion reactions according to the vesicle-specific phospholipid profile, and the organelle-specific components elaborated in ER and destined for the site. Thus, we came to a tentative conclusion that the delivery of ERgenerated transport vesicles to different membrane/organelles is initially dictated by the ER-determined membrane composition, followed by the action of the phospholipid-specific cytosolic PLA2 that initiates the vesicle-organelle specific fusion.
Inarguably, since PLA2 contributed to increase in mitochondrial LPG, the portion of the vesicular PG that during fusion with mitochondrial membrane was converted to LPG must have been in the outer leaflet of the

Figure 3. Lipid composition of purified mitochondria used for isolation of OMM, IMM and IMSC. The HPTLC was performed as described in detail under Figure 2. The analysis of separated lipids detected presence of cardiolipin (CL) and lysocardiolipin (LCL).
vesicle, and thus remained in the outer leaflet of OMM. Indeed, this notion was supported by the findings that upon further incubation with vesicles-free, transportactive cytosol (CC), the mitochondrial LPG was visibly reduced (Figure 5). Within 5 min of incubation, the level of LPG decreased discernibly, whereas under static conditions its contents in OMM was practically eliminated (Figure 6).
Accordingly, we concluded that the enzymes that aid fusion and reacylation of fusion-created LPG must represent a part of the mitochondrial coat and CC. One facilitates conditions enabling fusion, and the other eliminates the excess of harmful lysophospholipids and restores OMM integrity. Perhaps, the elimination of the excess of lysolipids may have been recognized by others [23,24] as mitochondria-specific fatty acid remodeling. If so, it could be that the mitochondria-specific PG and CL remodeling reflects the process and completion of transport from ER which is terminated by OMM LPG acylation.
As shown in Figure 4 the incubation of mitochondria with radiolabeled PG-containing transport vesicles afforded radiolabeled CL and LCL, which suggest that fusion process of PG containing vesicles is accompanied by the formation of mitochondria-specific CL and LCL. Therefore, we hypothesized that this is a direct outcome of vesicles cargo-induced synthesis of lipids in the OMM and IMM leaflets facing the inner space containing IMSC.
To determine the processes evoked by fusion of PG
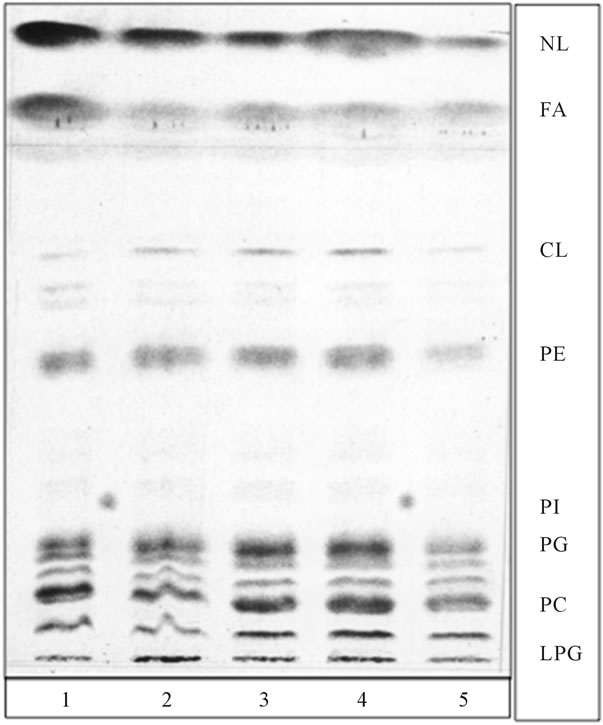
Figure 4. Fusion of radiolabeled PG transport vesicles, depicted in Figures 1(b) and 2, with cold mitochondria introduced radiolabeled lipids into mitochondria, generated radiolabeled CL and produced substantial increase in LPG. None of the preparations of purified mitochondria contained tracable amount of phosphatidylinositol (PI) or ceramides (Cer). The PG transport vesicles were derived from samples depicted in Figure 1(b) (lane 1 and 2), from samples subjected to fusion with ER, depicted in Figure 2 (lane 3, 4 and 5). Phosphatidylinositol (PI) isolated from ER–transport vesicles (Figure 1(a)) was spotted between lane 1 and 2, and 4.5. The thin layer chromatography plates and developing solvent systems were the same as in Figure 1, except that lipid extracts were applied in form of narrow elongated band.
vesicles with the mitochondrial membrane, and to determine whether the components of intra-mitochondrial contents are potentially involved in the deacylation of CL, the radiolabeled IMM membrane lipids were separated and the CL fraction subjected to incubation with IMSC (Figure 7). Indeed, the results of CL incubation with IMSC provided evidence that the IMSC contains CL-deacylating activity (Figure 7(b)).
As demonstrated in Figure 6 the OMM is practically free of CL, and hence by inference the IMM is enriched in CL. Considering the fact that OMM is ruptured and recovered in the form of membrane sheets, whereas IMM has the appearance of intact membrane “ghosts” [16], the major CL synthetic activity must be localized to the outer leaflet of IMM that is in physical contact with IMSC whose activity is dependent upon vesicular delivery of ER-produced cargo. This assumption is substantiated by the facts that IMSC supported the synthesis of CL
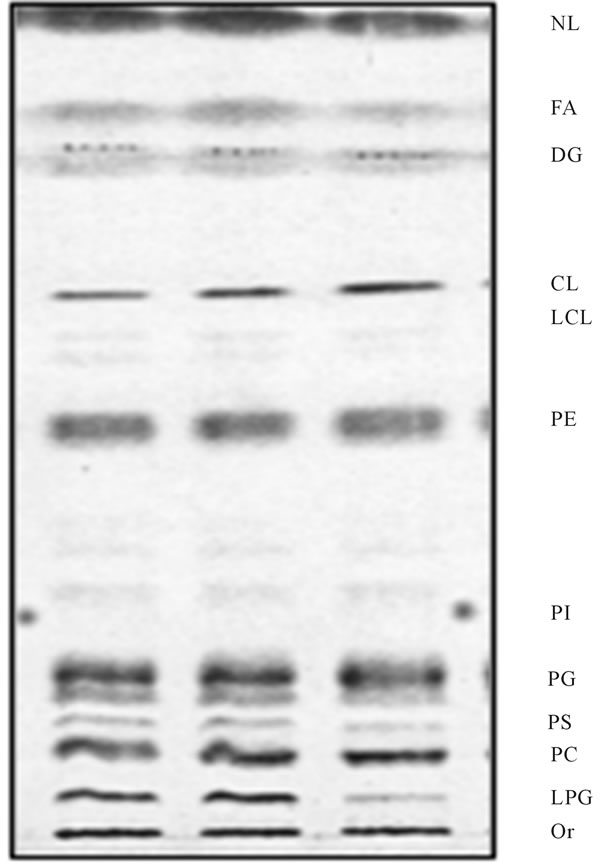
Figure 5. Reacylation of LPG generated in mitochondria during vesicular fusion. Following fusion experiment depicted in Figure 4, the aliquots of the mitochondria were subjected to further incubation with CC for up to 10 min at 4˚C and 37˚C, respectively. As evident in figure, the mitochondria incubated at 4˚C (middle lane) were displaying same lipid profile as nonincubated samples (left lane), whereas lipid profile of the samples recovered after incubation at 37˚C (right lane) contained substantially lesser amount of LPG. The lipid chromatography was the same as described for Figure 4.
(Figure 7(a)), whereas CC failed to produce the CL in either OMM or IMM. In our interpretation, this further implies that the synthesis of CL is dependent upon vesicular delivery of CL synthase from ER to the space between IMM and OMM. That seems to be the case, since relative amount of radiolabeled CL detected in aliquots withdrawn during fusion was substantially smaller than one detected in mitochondria after prolonged radiolabeling (Figures 3 and 5, respectively). That would suggest that IMM relies on CL synthese delivered in PG transport vesicles and therefore the appearance of radiolabeled CL (Figure 4) is connected to the ER transport vesicles fusion and delivery of radiolabeled substrate, and the enzymes that generate CL and LCL. Thus, we surmised that CL is generated in the inner mitochondrial membrane (IMM) and that formation of LCL is accomplished by the CL-specific lysophospholipase contained
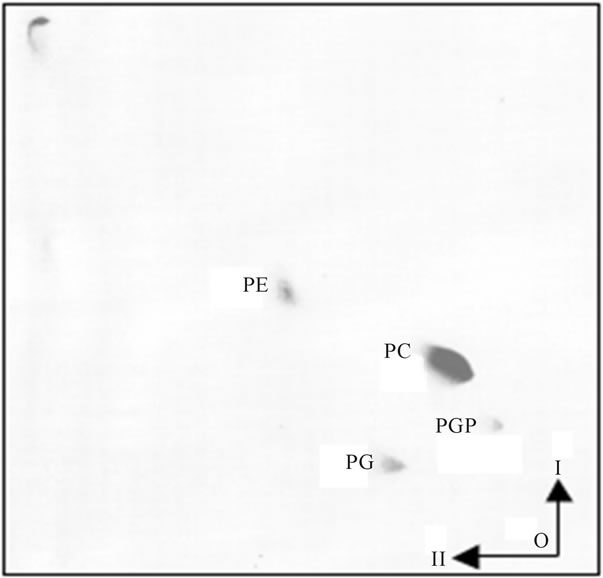
Figure 6. Lipid composition of OMM. The lipid analysis of purified OMM has not revealed detectible level of LPG. As displayed, the lipid composition of purified mitochondrial membranes showed demonstrable differences between OMM and IMM. As evident, the OMM lipid composition showed the presence of three major phospholipid components represented by PE, PC and PG. Also, traces of PGP were present, but LPG, LCL and CL were not detectable. The two-dimensional HPTLC was performed as described in legend for Figure 2. The preparation of OMM was accomplished by the procedure described in detail in Methods.
between OMM and IMM and that facilitates IMM permeability and transfer of CL synthase to matrix [23]. But normally, the analysis of mitochondrial membranes identifies rather small amount of LCL (Figure 3), and in the purified IMM not even traces of LCL are evident (Figure 7(a)). Yet, the results illustrated in Figure 7(b) clearly demonstrate that IMSC is enriched with cardiolipin deacylating enzymes. Therefore, the utter absence of LCL in purified IMM suggests that its level is determined by the CL-specific acyltransferases that reduce LCL contents in IMM, and stabilize IMM structure, while their effect is registered in CL fatty acyl uniformity [14,25].
In this study, we cannot assess whether the same transferases are eliminating LPG from IMM, since as is apparent in Figures 2, 4 and 5, the level of LPG in IMM remains rather high, and in the intact mitochondria, the IMM is not exposed to acyltransferase present in CC. Therefore, it is unlikely that LPG in IMM is reacylated by the IMSC or CC-derived acyltransferases. Though, with high degree of certainty, we may conclude that the CC-derived acyltransferase is involved in LPG elimination from OMM. As shown in Figure 5, following fusion, the LPG contained in mitochondria decreased upon further incubation with CC. On the other hand, the LPGspecific acyltransferase displayed poor activity toward
 (a) (b)
(a) (b)
Figure 7. Identification of radiolabeled CL in IMM and its susceptibility to IMCS deacylating enzyme activity. Radiolabeled CL depicted in mitochondria (Figure 3) was isolated from TLC plates and subjected to incubation with IMCS for 30 min at 37˚C. Following incubation the lipids were extracted from IMCS aliquots and subjected to separation in solvent mixture consisting of hexanes/diethyl ether/acetic acid 80/20/2. The results of the incubation with IMCS of CL depicted in panel (a) are shown in panel (b) which demonstrates release of fatty acids (FA).
LPC and LPIP suggesting that the cytosol-derived acyltransferases are specifically geared to eliminate fusion associated production of the organelle-specific lysolipids.
In conclusion, our results on transport vesicles generation and their affinity to intracellular organelles, and cell apical and basolateral membranes, allow us to deduce the major scheme of transport that in its entirety is dependent on ER-initiated synthesis of multiplicity of transport vesicles that at the same time but in varying quantities, are transported to cellular organelles and cell membranes. Based on the evidence acquired previously [2-4,7-11] and the data presented in this paper, there are two major routes of transport; one via Golgi apparatus and one to mitochondria. Golgi directed transport vesicles are enriched in PI and Cer lipid components, whereas those directed to mitochondria are discernible by the presence of PG. In Golgi, further maturation of Golgi-destined vesicles determines their membrane specificity and provides them with affinity to apical, basolateral cell membrane and to endosomes.
The mitochondria-directed PG-containing transport vesicles deliver protein derived from the nuclear DNA transcripts, of which CL synthase, CL-specific lysophospholipase and acyltransferases have been tentatively identified by following the lipid conversion during vesicular transport reflected in whole mitochondria and in the OMM and IMM. Since mitochondria-specific ERtransport vesicles and OMM are virtually-free of CL, and CC is not able to assemble CL in OMM or IMM, we further hypothesize that CL is mainly generated in IMM with the aid of protein cargo delivered from ER and released into the inner mitochondrial space (IMSC). Also, our study suggest that vesicular transport, delivery of the cargo and restitution of the specific organellar membrane is accomplished through a specific phospholipase action i.e. PC-specific PLA2 in fusion with Golgi, PIP-specific PLA2 in fusion with endosomes and apical and basolateral epithelial membrane, and PG-specific PLA2 in fusion with mitochondria.
It seems plausible that as the fusion of transport vesicles is accomplished through generation of lysolipids, and the reaction is completed through elimination of these membrane destabilizing lipids. Therefore, the purified OMM and IMM are virtually free of lysolipids, whereas in the fusion experiments their presence in membranes is demonstrable. Same phenomenon seem to operate in the processes associated with LCL generation and its elimination in the IMM. After LCL-induced destabilizing of IMM that provide access gate for nuclear DNA products to mitochondrial matrix, the LCL is eliminated. These results confirm that the cellular transport is achieved in similar but highly specific mode, by producing membrane destabilizing specific lysophospholipids that contribute to a local opening of the membranes, membranes fusion, and cargo release.
The results presented above allow us to advance further our evidence-driven hypothesis that ER generates a wide range of cell-specific transport vesicles that deliver organelle-specific membrane and cargo, and thus repair and reinstitute cell organelles and membranes in highly controlled fashion. The transport vesicles that are aimed to fulfill the restoration processes in mitochondria have mitochondria-characteristic lipid composition, and deliver nuclear DNA translation products to the inner and to matrix mitochondrial space in order to facilitate CL synthesis and thus preserve CL-requiring phosphorylative oxidation to produce cell sustaining energy.
4. DISCUSSION
The initial concept of cellular transport built on the results generated from the investigations of Golgi transport vesicles remains unchanged [5,6,26-28]. And, after 20 years of research dedicated to the topic, the findings that the biogenesis and the attributes of the vesicles are gained in ER are not incorporated into the model. Instead, the Golgi vesicles-associated proteins dominate further investigative inquires that concentrates on the protein exclusivity in the process [29-34]. In the theory enforce, the biomembrane of the cell and the variety of cell organelles, although recognized for their uniqueness and need to be restored with utmost fidelity and timeliness, are loosely interpreted as inconsequential scaffold for protein. The preoccupation with a potential of the initially recognized in Golgi transport-associated proteins and proposed hidden meaning of their minute structural details leads to tortuous explanations of their function in other cellular transporters and, obscures the significance of the features that in major way contribute to a distinctiveness of the process that is initiated in ER [2-4,7-10]. Consequently, the maintenance of the vesicle framework which determines its true destination is minimized by suggesting that the transporters are serving as mere containers delivering proteins and returning for the next round of transport [21]. In an essence, the initially conceived theory, although commends dominance and thus gained many followers, cannot explain how membrane specificity is maintained, how the organelles variable restitution is accomplished, how the perfect balance between membrane lipids and membrane proteins is achieved, or how the cellular or organellar membrane proteins are so precisely intercalated into the specific lipid scaffold.
In contrast, our investigations reveal a coherent picture of the cellular repair and restitution in which all facets of the specific transport rely on the assembly of ER transport vesicles and their precisely maintained lipid and protein composition. Cellular vesicular transport is initiated in the ER with active translation processes [3]. If viable cytosol of one type of cells is substituted with other cells cytosol, the resultant vesicles are modified. Moreover, if cytosol is treated with RNase, neither vesicles synthesis nor the nuclear transport is observed [4]. The ER generated transport structures contain newly translated membrane and cargo proteins, and morphologically appear similar to those initially observed in Golgi [35]. But upon biochemical analysis, we found that ER vesicles differ substantially from those generated by Golgi. The newly translated proteins transported to Golgi for their posttranslational modifications are encased in the vesicles consisting of newly synthesized lipids represented by PC, PE, PI and Cer [2,9-11]. In Golgi, their immature membranes that undergo protein posttranslational modification are also augmented with sphingomyelin (SM), glycosphingolipids (GSL) and phosphatidylinositol phosphates (PIPs) [9,11]. The membrane lipid modification is destination specific; the vesicles transporting cargo to apical membrane gain SM, GSL and PI3P through alteration of PC, Cer and PI, respectively, those destined to endosomes gain SM and PI4P, whereas those transporting to basolateral membrane come to have SM and PIP2 [3,9,11]. In our interpretation, the acquired differences signify predetermined by ER lipid environment for the translated transcripts of ER transport vesicles membrane and their cargo, and feature characteristic lipid components of the sites of their delivery. By the virtue that in our investigations the newly synthesized ER transport vesicles were lipid radiolabeled, we were able to establish that the release of protein cargo and the en bloc vesicular membrane incorporation into the organelle has occurred [2,3,9,11]. Thus, contrary to broadcasted interpretation, the ER assembled transport vesicles were not taking numerous transport rounds within the cell, but were incorporated into specific cell membrane and thus renewed the segment of a such membrane. Consistent with our findings-based postulation, the vesicles maturing in Golgi are not identical but are modified in concurrence to replace the specific membrane fragment of apical epithelial membrane, endosomal and basolateral membrane and, as well not yet investigated other cellular organelles and sites, to renew them and deliver specific cargo [3,8-10]. Therefore, the great majority of ER vesicles consisting of still unrefined cellular products and requiring Golgi-specific posttranslational modification, first carry common affinity for Golgi membrane, whereupon the transfer, their cargo and their membranes undergo critical processing into different sets of transporters. Specifically, these vesicles attain particular lipid markers (GSL, SM, PIP) that determine their final destination [9-11]. Relevantly, though, none of the vesicular membrane modifications resemble ER composition, or the vesicles exhibit any affinity for ER, that would dictate their return to ER for new cargo. In our opinion, the results provide strong support to a concept that ER produces, at the same time but at different quantitative demand a gamut of cellular vesicular transporters of which some require Golgi-mandated destination-specific modification. From there, with their particular cargo and in precisely structured replacement of the portion of cellular or organellar membrane, the vesicles reach the site intended [2,3,9-11].
Along the same line of thinking the connection between nuclear membranes biogenesis and intranuclear transport of cytosolic proteins was deciphered [4,7]. The investigation of cytosolic protein transfer to nucleus, aided by its affinity to PIPs, demonstrated the involvement of phosphoinositides in the transport. This allowed us to speculate that in the nuclear-ER membrane continuum the PIPs phosphorylation-dephosphorylation cycle is engaged in the transport, synthesis and the renewal of ER/nuclear membranes, deposition of cytosolic protein in the nucleus, and the exodus of the cytosolic protein signal-ignited transcripts of mRNA to cytosol. The pulse chase study of the nuclear inner membrane (INM) and the outer nuclear membrane (ONM) PIPs has determined the existence of the cycle and re-immergence of the membrane containing ER-specific phospholipids containing dephosphorylated PI core. This provided an explanation for the absence of PIPs in ER and yet the uninterrupted seamless continuum between nuclear and ER membranes [4]. Undeniably, the membrane lateral movement and the exposure to intranuclear products replenished cytosolic transcripts of RNA that dictated translation of the specific protein and formation of the specific membranes. The seamless connection between nuclear PIPs cycle and ER membrane genesis gave credence to the signal-triggered processes that culminate in continuous and faithful restoration of cell characteristic protein and lipid components and functions, but without need for the detached vesicular delivery.
As our investigations generated support to the idea that the transport vesicles are not mere containers for protein delivery but a specific apparatus to distinctively restore cell’s membranes, the means and the course for restoration of mitochondrial proteins and membranes remained unknown. By connecting the nuclear to ER to Golgi transport events, we could not envision restitution of the mitochondrium. Clearly, the ER transport vesicles directed to Golgi were not containing mitochondria-specific lipids and that negated the Golgi path for transfer of nuclear transcripts’ protein products to mitochondria. The current hypothesis, suggesting that distinct domains of ER mediate interaction with mitochondria and such contacts provide transport hubs for calcium and proteins encoded by nuclear DNA [36-43] does not explain how the accidental points of contact between two organelles could restore mitochondrial membranes composition and deliver in timely fashion mitochondria-specific proteins required in the space between inner and outer mitochondrial membrane and by matrix.
In the studies presented in this paper, we provide evidence that supports a direct vesicular transport from ER to mitochondrium. These vesicles were first identified in the experiments with BFA [20]. The synthesis of transport vesicles in the presence of BFA, the inhibitor of ER-Golgi transport, afforded generation of the vesicles which did not display affinity with Golgi membranes, consisted of lipids enriched with phosphatidylglycerol (PG), phosphatidylcholine (PC) and phosphatidylethanolamine (PE) and were virtually free of PI, sphingomyelin (SM) and glycosphingolipids (GSL). At the time, the results of the findings were interpreted as BFA-induced modification of ER vesicles biogenesis [20]. However, identical vesicles were detected in the experiments without BFA, and hence we reasoned that they must be pointing toward still undetermined cellular function. As shown in the results, the fraction of ER transport vesicles that remained in the cytosol when normal transport to Golgi was accomplished, and those identified in BFA experiments, consisted of PG, PC and PE, while the Cer and PI were not detectable. Moreover, the mucosal epithelialand hepatocytes-derived PG vesicles did not contain apomucin or albumin cargo, respectively. Thus, the absence of such cargo, allowed us to suggest that these vesicles must carry products necessary for other than Golgi-connected sites, the sites that require PG and are not involved in the secretion of mucin or albumin.
PG, although seemingly common phospholipid in ER membranes where its de novo synthesis is initiated with formation of CDP-DG from PA is an important precursor for the synthesis of cardiolipin (CL), a polyglycerylphospholipid that is required for the activity of a large number of enzymes residing in mitochondrial core and the membrane lipids found only in mitochondrial membranes. However, even the ER vesicles that showed affinity to mitochondrial membrane did not contain detectible amount of CL. Therefore, we concluded that the PG enriched vesicles must deliver PG containing membranes and ER-translated protein cargo to assemble CL in the mitochondrium [40,42-47]. Since analysis of the mitochondrial membranes incubated with PG enriched vesicles revealed the presence of LPG, we surmised that this is the immediate consequence of fusion reaction that involves deacylation of PG [45,46]. Just as formation of LPC in Golgi and LPIP in apical epithelial membranes, the formation of LPG apparently precedes and facilitates incorporation of the vesicular membrane into mitochondrial OMM when releasing ER-produced cargo between OMM and IMM. Formation of LPG and the presence of LPG-specific acyltransferase in ER [46], suggest that the enzyme must be an integral part of the PG transport vesicles that upon fusion and LPG formation, reacylates it to PG. This process, thus could explain the mitochondrial PG fatty acid remodeling and elimination of the lysophospholipids from the OMM. The fact that the lysophosphatidyl acyltransferase has no significant acyltranferase activity toward LPC, LPE, LPS and LPI strongly suggest that the processing of LPG to PG is mitochondria-specific and occurs in the outer OMM leaflet where the specific fatty acyl CoA can be incorporated [37].
Neither in the vesicles identified in BFA experiments, nor the vesicles remaining in cytosol after delivery of cargo to Golgi, the cardiolipin (CL) specific marker of mitochondrial lipids was detected. Hence, we surmised that CL synthase is not present in the cell cytosol and also is not active in the mitochondrial transport vesicles. If it was, the vesicles enriched with PG would also contain CL, since the CDP-DG substrate is available in ER and used to generate PI in ER vesicles destined for transport to Golgi. Moreover the CDP-DG could not reside on cytosolic leaflet of mitochondrial vesicles or the mitochondria, since PI was not identified in OMM. Thus, the findings suggested that CL synthase is delivered to mitochondria in PG-containing mitochondria-specific transport vesicles and produces CL in the membrane leaflets facing the space generated between OMM and IMM [48].
The facts that RNase and proteinase K treatments of the cytosol abolish transport, support the most significant premise of cellular activity; one, ceasing generation of transport vesicles, and the other curtailing vesicles fusion. As we demonstrated earlier [11,22], the vesicles fusion with organelles is aided through cytosolic phospholipases A2. The fact that incorporation of vesicle membrane lipids into mitochondrium is associated with production of LPG demonstrated that this event is aided by the PGspecific mitochondria-associated phospholipase A2.
These initial results allowed us to suggest that ER translated proteins are delivered to mitochondrium in the mitochondria-specific transport vesicles. However, the vesicles produced in the ER do not supply mitochondrial membranes with cardiolipin (CL). Thus, either CL is synthesized only in the IMM and the CL synthase is delivered in mitochondrial cargo from the ER and released into the space between OMM and IMM, or by the mitochondrial matrix. Our results on lipid composition of OMM and IMM show that only traces of CL are detected in OMM, whereas the IMM contains substantial fraction of CL. Also, since cell cytosol does not support the synthesis of CL, it is apparent that CL synthase is contained within OMM and IMM, and must be delivered with vesicular cargo that does not spill into the cytosolic environment.
In the studies documenting the fusion of ER transport vesicles with Golgi we determined that PC-specific PLA2 was aiding fusion of the membranes [11,22], whereas fusion of Golgi vesicles with apical membrane was assisted by PIP-specific PLA2 [8,11,22]. In the case of mitochondria-directed transport vesicles, possibility exists that a similar mechanism is engaged. The PG-specific PLA2 produces lysolipids which contribute to membranes local lysis and the resulting destabilization allows incorporation of the transport vesicle membrane into OMM. While analysis of IMM also revealed the presence of LCL it is difficult to speculate whether in turn, the LCL contributes to the destabilization of the IMM resulting in accessibility of CL synthase to matrix and its lipid substrates. The evidence on CL fatty acid remodeling, however, suggests that PG and CL undergo PLAspecific deacylation and mitochondria-specific acylation, evidently through substitution of the fatty acids produced by mitochondrial FA synthase in the leaflet facing matrix [23,39,40].
In retrospect, the initial study on cellular transporters utilized Golgi as the earliest and the primary site that produced transport vesicles [5]. Indeed, by omitting initial stage of vesicles biogenesis, the mitochondrial portion of transport vesicles was overlooked, while the Golgi vesicles appearance, judged by the presence of glycosylated secretory cargo, failed to detect the transporters destined for intracellular organelles or basolateral portion of the cell membrane. It is possible that the fraction of the transporters, meant for other than apical membrane destination, is still assumed as the vesicles returning to ER for new apical cargo or as portion that was not utilized under the conditions of the experiment. This, however, could be only established by characterization of the remaining vesicles. In our studies, by utilizing nonradioactive ER membranes and radiolabeled transport vesicles, we could eliminate such turn of events and establish that the entire spectrum of so far characterized vesicular transporters does not return to ER. The prove of that is ascertained by the evidence documenting that vesicular membranes lipids (SM, GSL, PIPs) were not found in the ER. That alone, provides sufficient prove that transport vesicles do not return for new cargo.
Considering results of our earlier studies, and combining past as well as the present findings, we clearly see that BFA, as suggested earlier [11,20] impacts the restitution of Golgi membranes, and this impacts Golgi structure as well as its ability to process vesicles transiting Golgi and delivering cargo to cell membrane and to other organelles. It is not surprising that proteins from the cytosol that under normal conditions associate with Golgi, in the presence of BFA that prevents restitution of Golgi, dissociate due to lack of the affinity with modified Golgi membrane. Hence, we speculate that BFA may be impacting translation of proteins and membranes biogenesis that transit through Golgi network, meanwhile the mitochondria-destined vesicles appear not affected.
Thus, the nuclear signal-initiated template, that is released to cytosol in the form of signal-determined array of RNAs, dictates production of the specific variety of transport vesicles. In comparison, the presently supported hypothesis based on the associated protein function cannot withstand the requirements that are obligatory for cohesive and cell genuine restitution. Under the conditions described in the latter concept, it is not achievable that multiplicity of membranes, their receptors, and the integral proteins would be synthesized and precisely embedded. Such integration can only be accomplished if the translation and membrane synthesis is synchronized in time and space [2-4].
While the initial impression of the nuclear membranes restitution seemed to set apart the processes contributing to the transport of cytosolic proteins to nucleus, the fact that nuclear membrane represents a portion of endoplasmic continuum provided explanation for lack of vesicular transport. As our previous studies revealed, the continued ER membrane that envelopes nuclear contents is restored by progressive lateral movement of ER membrane that is evoked by the synthesis of membrane lipids and specific modification of the PI into PIPs serving as cytosolic protein receptors and transporters. The lipid synthesis-induced movement through one nuclear pore provides nucleus with newly translated nuclear proteins and the renewal of INM, while through the other pore site emerges the newly refurbished INM exporting nuclear components transcribed in response to the entering signal from cytosolic site. In our interpretation, the lateral movement of the nuclear portion of the ER continuum endows nucleus with a steady influx of cytosolic signal proteins, and the ER with steady influx of the membrane that is conditioned to generate vesicles according to nucleus delivered template [13,18,36,49].
Based on lipid analysis of the ER, transport vesicles, and the mitochondrial membranes, we suggest that all organelles in the cell are repaired and renewed in a highly specific organized way that is initiated in the ER by the nucleus-determined signals which create new fragments of organelle specific membranes that enclose organelle-specific cargo [2-4,7-11,49]. Indeed, in the studies presented here we documented that mitochondria are rebuilt and replenished with the ER translated protein through generation of transport vesicles which are directly delivered to mitochondria, while other vesicles are prepared for their final destination in Golgi.
![]()
![]()
REFERENCES
- Croze, E.M. and Morre, D.J. (1984) Isolation of plasma membrane, Golgi apparatus, and endoplasmic reticulum from single homogenates of mouse liver. Journal of Cellular Physiology, 119, 46-57. doi:10.1002/jcp.1041190109
- Slomiany, A., Sano, S., Grabska, M., Yamaki, K. and Slomiany, B.L. (2004) Gastric mucosal cell homeostatic physiome. Critical role of ER-initiated membrane restitution in the fidelity of cell function renewal. Journal of Physiology and Pharmacology, 55, 837-860.
- Slomiany, A. and Slomiany, B.L. (2010) Cell membranes composition is defined in ER and their restitution proceeds by en bloc fusion of ER generated transport vesicles. Health, 2, 1444-1454. doi:10.4236/health.2010.212214
- Slomiany, A. and Slomiany, B.L. (2011) Transformations of phosphatidylinositol phosphates in the outer and inner nuclear membrane are linked to synthesis and restitution of cellular membranes. Health, 3, 187-199. doi:10.4236/health.2011.34035
- Rothman, J.E. (1994) Mechanisms of intracellular protein transport. Nature, 372, 55-63. doi:10.1038/372055a0
- Lemmon, M.A. (2008) Membrane recognition by phospholipids binding proteins. National Review of the Molecular Cell Biology, 9, 99-111. doi:10.1038/nrm2328
- Slomiany, A., Grabska, M. and Slomiany, B.L. (2006) Homeostatic restitution of cell membranes. Nuclear membrane lipid biogenesis and transport of protein from cytosol to intranuclear spaces. International Journal of Biological Sciences, 2, 216-226. doi:10.7150/ijbs.2.216
- Slomiany, A. and Slomiany, B.L. (2003) Lipidomic processes in homeostatic and LPS-modified cell renewal cycle. Role of phosphatidylinositol 3-kinase pathway in biomembrane synthesis and restitution of apical epithelial membrane. Journal of Physiology and Pharmacology, 54, 533-551.
- Slomiany, A., Grabska, M., Piotrowski, E. and Slomiany, B.L. (1994) Intracellular processes associated with vesicular transport from endoplasmic reticulum to Golgi and exocytosis. Archives of Biochemistry and Biophysics, 310, 247-255. doi:10.1006/abbi.1994.1164
- Slomiany, A., Grzelinska, E., Grabska, M. and Slomiany, B.L. (1992) Intracelular processes associated with glycoprotein transport and processing. Archives of Biochemistry and Biophysics, 298, 167-175. doi:10.1016/0003-9861(92)90108-9
- Slomiany, A., Nowak, P., Piotrowski, E. and Slomiany, B.L. (1998) Effect of ethanol on intracellular vesicular transport from Golgi to the apical membrane. Role of phosphatidylinositol 3-kinase and phospholipase A2 in Golgi transport vesicles association and fusion with the apical membrane. Alcohol, Clinical and Experimental Research, 22, 167-175. doi:10.1111/j.1530-0277.1998.tb03634.x
- Neupert, H. and Herrmann, J.M. (2007) Translocation of proteins into mitochondria. Annual Review of Biochemistry, 76, 723-749. doi:10.1146/annurev.biochem.76.052705.163409
- Becker, T., Gebert, M., Pfanner, N. and van der Lannn, M. (2009) Biogenesis of mitochondrial membrane proteins. Current Opinions in Cell Biology, 21, 484-493. doi:10.1016/j.ceb.2009.04.002
- Osman, C., Voelker, D.R. and Langer, T. (2011) Making heads or tails of phospholipids in mitochondria. Journal of Cell Biology, 192, 7-16.
- Claypool, S.M., Octay, Y., Boontheung, P., Loo, J.A. and Koehler, C.M. (2008) Cardiolipin defines the interactome of the major carrier protein of mitochondrial inner membrane. Journal of Cell Biology, 182, 937-950. doi:10.1083/jcb.200801152
- Parsons, D.F., Willims, G.R. and Chance, B. (1966) Characteristics of isolated and purified preparations of the outer and inner membranes of mitochondria. Annals of the New York Academy of Sciences, 137, 643-666. doi:10.1111/j.1749-6632.1966.tb50188.x
- Cobon, G.S., Crowfoot, D. and Linnane, W. (1974) Biogenesis of mitochondria. Phospholipid synthesis in vitro by yeast mitochondrial and microsomal fractions. Biochemistry Journal, 144, 265-275.
- Gohil, V.M. and Greenberg, M.L. (2009) Mitochondrial membrane biogenesis: Phospholipids and proteins go hand in hand. Journal of Cell Biology, 184, 469-472.
- Klausner, R.D., Donaldson, J.G. and Lippincott-Schwartz, J. (1992) Brefeldin A: Insights into the control of membrane traffic and organelle structure. Journal of Cell Biology, 116, 1071-1080. doi:10.1083/jcb.116.5.1071
- Slomiany, A., Grabska, M., Slomiany, B.A., et al. (1993) Intracellular transport, organelle biogenesis and establishment of Golgi identity: Impact of Brefeldin A on the activity of lipid synthesizing enzymes. International Journal of Biochemistry, 25, 891-901. doi:10.1016/0020-711X(93)90245-A
- Idone, V., Tam, C. and Andrews, N.W. (2008) Two-way traffic on the road to plasma membrane repair. Trends in Cellular Biology, 18, 552-559. doi:10.1016/j.tcb.2008.09.001
- Slomiany, A., Grzelinska, E., Yamaki, K. and Slomiany, B.L. (1992) Function of intracellular phospholipase A2 in vectorial transport of apoproteins from ER to Golgi. International Journal of Biochemistry, 24, 1397-1406.
- Saini-chohan, H.K., Holmes, M.G. and Chicco, A.J., et al. (2009) Cardiolipin biosynthesis and remodeling enzymes are altered during development of heart failure. Journal of Lipid Research, 50, 1600-1608. doi:10.1194/jlr.M800561-JLR200
- Helmy, F.M. (2006) Cardiolipin, its preferential deacylation in mammalian myocardia. Mini review and chromatographic-computational analysis. Acta Chromatographica, 17, 9-19.
- Cao, J., Liu, Y., Lockwood, J., Burn, P. and Shi, Y. (2004) A novel cardiolipin-remodeling pathway revealed by a gene encoding an endoplasmic reticulum-associated acylCoA: Lysocardiolipin acyltransferase (ALCAT1) in mouse. The Journal of Biological Chemistry, 279, 31727-31734. doi:10.1074/jbc.M402930200
- Beck, R., Prinz, S., Distellkoter-Bechart, P., et al. (2011) Coatomer and dimeric ADP ribosylation factor 1 promote distinct steps in membrane scission. Journal of Cell Biology, 194, 765-777.
- Sarrl, E., Sicart, A., Lazaro-Dieguez, F. and Egea, G. (2011) Phospholipid synthesis participates in the regulation of diacylglycerol required for membrane trafficking at Golgi complex. The Journal of Biological Chemistry, 286, 28632-28643. doi:10.1074/jbc.M111.267534
- Ma, C., Agrawal, G. and Subramani, S. (2011) Peroxisome assembly: Matrix and membrane protein biogenesis. Journal of Cell Biology, 193, 7-16. doi:10.1083/jcb.201010022
- Sudhof, T.C. and Rothman, J.E. (2009) Membrane fusion: Grappling with SNARE and SM proteins. Science, 323, 474-477. doi:10.1126/science.1161748
- Glick, B.S. and Nakano, A. (2009) Membrane traffic within the Golgi apparatus. Annual Review of Cell Development Biology, 25, 113-132. doi:10.1146/annurev.cellbio.24.110707.175421
- Chapman, E.R. (2008) How does synaptotagmin trigger neurotransmitter release? Annual Review of Biochemistry, 77, 615-641. doi:10.1146/annurev.biochem.77.062005.101135
- van Meer, G., Halter, D., Sprong, H., Somerharju, P. and Egmond, M.R. (2006) ABC lipid transporters: Extruders, flippases, or flopless activators? FEBS Letters, 580, 1171- 1177. doi:10.1016/j.febslet.2005.12.019
- Kawano, M., Kumagai, K., Nishijima, M. and Handa, K. (2006) Efficient trafficking of ceramide from endoplasmic reticulum to the Golgi apparatus requires VAMPassociated protein interacting FFAT motif of CERT. Journal of Biological Chemistry, 281, 30279-30288. doi:10.1074/jbc.M605032200
- Mellman, I. and Nelson, W.J. (2009) Coordinated protein sorting, targeting and distribution in polarized cells. National Review of the Molecular Cell Biology, 9, 833-845. doi:10.1038/nrm2525
- Slomiany, A., Grzelinska, E.,Kasinathan, C., et al. (1992) Biogenesis of endoplasmic reticulum transport vesicles transferring gastric apomucin from ER to Golgi. Experimental Cell Research, 201, 1669-1682. doi:10.1016/0014-4827(92)90280-L
- Simmen, T., Lynes, E.M., Gesson, K. and Thomas, G. (2010) Oxidative protein folding in the endoplasmic reticulum: Tight links to the mitochondria-associated membrane (MAM). Biochimica et Biophysica Acta (BBA): Biomembranes, 1798, 1465-1473. doi:10.1016/j.bbamem.2010.04.009
- Berridge, M.J. (2002) The endoplasmic reticulum: a multifunctional signaling organelle. Cell Calcium, 32, 235- 249. doi:10.1016/S0143416002001823
- Nie, J., Hao, X., Chen, D., et al. (2010) A novel function of the human CLS1 in phosphatidylglycerol synthesis and remodeling. Biochimica et Biophysica Acta (BBA): Biomembranes, 1801, 438-445. doi:10.1016/j.bbalip2009.12.002
- Houtkooper, R.H. and Vaz, F.M. (2008) Cardiolipin the heart of mitochondrial metabolism. Cellular and Molecular Life Sciences, 65, 2493-2414. doi:10.1007/s00018-008-8030-5
- Zachman, D.K.K., Chicco, A.J., McCune, S.A., et al. (2010) The role of calcium-independent phospholipase A2 in cardiolipin remodeling in the spontaneously hypertensive heart failure rat heart. Journal of Lipid Research, 51, 525-534. doi:10.1194/jlr.M000646
- Darios, F., Ruiperez, V., Lopez, I., et al. (2010) Alphasynuclein sequesters arachidonic acid to modulate SNAREmediated exocytosis. EMBO Reports, 11, 528-533. doi:10.1038/embor.2010.66
- Chen, D., Zhang H.-Y. and Shi, Y. (2006) Identification and functional characterization of hCLs1, a human cardiolipin synthase localized in mitochondria. Biochemical Journal, 398, 169-176. doi:10.1042/BJ20060303
- Osman, C., Haag, M., Wieland, F.T., Brugger, B. and Langer, T. (2010) A mitochondrial phosphatase required for cardiolipin biosynthesis: The PGP phosphatase Gep4. The EMBO Journal, 29, 1976-1987. doi:10.1038/emboj.2010.98
- Gebert, N., Joshi, A.S., Kutik, S., et al. (2009) Mitochondrial cardiolipin involved in outer-membrane biogenesis: Implications for Barth syndrome. Current Biology, 19, 2133-2139.
- Nie, J., Hao, X., Chen, D., et al. (2010) A novel function of the human CLS1 in phosphatidylglycerol synthesis and remodeling. Biochimica et Biophysica Acta, 1801, 438- 445. doi:10.1016/jbbalip.2009.12.002
- Yang, Y., Cao, J. and Shi, Y. (2004) Identification and characterization of a gene encoding human LPGAT1, an endoplasmic reticulum-associated lysophosphatidylglycerol acyltransferase. The Journal Biological Chemistry, 279, 55866-55874. doi:10.1074/jbc.M406710200
- Kawasaki, K., Kuge, O., Chang, S.-C., et al. (1999) Isolation of a chinese hamster ovary (CHO) cDNA encoding phosphatidylglycerolphosphate (PGP) synthase, expression of which corrects the mitochondrial abnormalities of a PGP synthase-defective mutant of CHO-K1 cells. The Journal of Biological Chemistry, 274, 1828-1834. doi:10.1074/jbc.274.3.1828
- Chen, D., Zhang, X.-U. and Shi, Y. (2006) Identification and functional characterization of hCLS1, a human cardiolipin synthase localized in mitochondria. Biochemical Journal, 398, 169-176. doi:10.1042/BJ20060303
- Giorgi, C., De Stefani, D., Bononi, A., Rizzuto, R. and Pinton, P. (2009) Structural and functional link between the mitochondrial network and the endoplasmic reticulum. International Journal of Biochemistry and Cell Biology, 41, 1817-1827. doi:10.1016/j.biocel.2009.04.010