American Journal of Molecular Biology
Vol. 2 No. 4 (2012) , Article ID: 23996 , 12 pages DOI:10.4236/ajmb.2012.24037
An engineered Phlebia radiata manganese peroxidase: Expression, refolding, purification and preliminary characterization
![]()
1Department of Chemistry and Biochemistry, School of Life Sciences, University of Sussex, Brighton, UK
2Department of Biochemistry, Faculty of Basic Medical Sciences, University of Uyo, Uyo, Nigeria
Email: Usen_mboso@yahoo.com
Received 21 July 2012; revised 21 August 2012; accepted 20 September 2012
Keywords: Phlebia radiata; Peroxidase; Manganese Peroxidase; Over-Expression; Recombinant
ABSTRACT
Manganese peroxidases (MnPs) are interesting enzymes in protein engineering, aimed at maximizing industrial bioprocesses such as lignin degradation and biofuel production. cDNA of the secreted shorttype of MnP from Phlebia radiata (Pr-MnP3) has been successfully engineered and amplified by polymerase chain reaction (PCR). Five mutant genes (E40H, E44H, E40H/E44H, D186H and D186N) of recombinant Phlebia radiata MnP3 (rPr-MnP3) were generated. The wild-type and the mutant genes were expressed in Escherichia coli (W3110 strain) and the resultant body proteins were lysed, purified and refolded into active enzymes. 6% - 7% recovery of pure and fully active rPr-MnP3 for wild-type and mutants were obtained and the availability of rPr-MnP3 enzymes will greatly facilitate its structure-function relationships studies. rPr-MnP3 mass was characterised using SDS-PAGE and MALDI-TOF mass spectrometry. Molecular weight of both the wild-type and mutant rPr-MnP3 enzymes was approximately 36 kDa. This paper describes the spectral characterization of the wild-type and mutant rPr-MnP3 enzymes with very close similarities; substantially high spin haem enzymes. Therefore we report the engineering, cloning, expression, refolding/activation of MnP3 genes and preliminary characterization of the wild-type and mutant Phlebia radiata MnP3 enzymes.
1. INTRODUCTION
Phlebia radiata functions as a wood-decaying, white-rot causing corticoid basidiomycete capable of selective and effective degradation of natural and synthetic lignins, lignin modeled compounds and various xenobiotics [1-5]). As a white-rot basidiomycetous and wood-decaying fungus, P. radiata expresses and secretes multiple extracellular lignin-modifying, class II type of haemcontaining peroxidases [6] including three isoenzymes of manganese peroxidase (MnP, EC 1.11.1.13) and three lignin peroxidases (LiP, EC 1.11.1.14), as well as two multicopper laccases (EC 1.10.3.2) [7-10].
Manganese peroxidase (MnP) is an extracellular oxidoreductase, a class II fungal haem-containing peroxidases belonging to the plant peroxidase-like protein superfamily [11]. MnP was first found in the white-rot fungus Phanerochaete chrysosporium [12,13] and there-after in most of the wood lignin and litter-decaying basidiomycetes [10,14].
MnP occurs as a series of isoenzymes encoded by a large number of genes [15-18]. Basically, three different types of fungal MnP enzymes are structurally characterized [10] long type of MnPs found in P. chrysosporium and in P. radiata (Pr-MnP2 enzyme) [19,20], short type of MnPs which are frequently detected as gene models in fungal genomes, but first isolated and described in P. radiata (Pr-MnP3) [8,20]. The third type of fungal MnP is the versatile peroxidase (VP, 1.11.1.17) which is structurally similar to the short-MnPs and enzymatically displaying hybrid activity for both Mn2+ ions and veratryl alcohol [21].
The physiological function of MnP is that of oxidation of Mn2+ ions to Mn3+ which is chelated by dicarboxylic acid leading to oxidative depolymerisation of phenolic compounds such as lignin [7,22,23]. Crystal structure of P. chrysosporium manganese peroxidase-H4 (MnPH4) clearly presents one Mn ion binding site in the vicinity of the second haem propionate within a short channel to the C-terminal surface of the protein [24]. Mn2+ is hexacoordinated including the haem propionate and three acidicconserved amino-acid residues (E, E, D) forming the Mn-binding site. This site as demonstrated by 3D homology modelling is present in Pr-MnP3 and Pr-MnP2 [20]. MnP has also been shown to depolymerise lignin in vitro [18]. Moreover, this enzyme mediates initial steps in the degradation of high-molecular weight lignin [12] and has been found in many wood and lignin-decomposing white-rot fungi [25].
The native enzymes of Phlebia radiata have been purified in liquid medium [26-28] and from solid-state fermentation cultures but insufficient to be fully characterized [3]. However, individual enzymes have not previously been expressed in recombinant form to allow full characterization. Genes encoding different MnP isozymes of P. radiata have been isolated [29] and structurally, Pr-MnP3 protein resembles VP and LiP, having a shorter C-terminal tail.
The production of recombinant proteins and their variants by site-directed mutagenesis is a pre-requisite for structure-function studies. Genes encoding peroxidase from different organisms have already been cloned and homologously or heterologously expressed in different hosts. The first active haem peroxidase of the plant peroxidase-like superfamily to be over-expressed heterologously in a bacterial host was a class I type haem peroxidase, the yeast mitochondrial Cytochrome c Peroxidase (CcP) [20]. Since CcP expression, Escherichia coli has been used to express many different haem-containing peroxidises: Horseradish peroxidase (HRP C) [30], lignin peroxidase (Pr-LiPH8) [31,32], Coprinopsis cinerea Peroxidase (CiP) [33], Versatile Peroxidase (VP) [34], long type of manganese peroxidase (Pr-MnPH4) [35]. Heterologous recombinant expression systems for the PrMnPs, such as E. coli, insect cell, yeast and filamentous ascomycetous fungi [16,36,37] have been established [32, 38], hence the structure-function studies of the long-type MnP enzymes [39-41].
All previous attempts to over-express MnP in either P. chrysosporium or alternative host have resulted in low yields [38,42,43]. Equally, low levels of active MnP were detected in the growth medium in the baculovirus expression system [44]. However, high-level expression of plant and fungal haem peroxidases have been achieved in Escherichia coli and the recombinant proteins usually accumulate in an insoluble form in cytoplasmic inclusion bodies.
This work is aimed at manipulating the MnP3 gene of P. radiata to allow high-level expression in E. coli, generation of MnP3 mutants of the Mn (II) binding site by site-directed mutagenesis with the hope of allowing the specificity of the site to be modulated with respect to both Mn2+ and other metals. The refolding and purification of the wild-type and mutant rPr-MnP3 variants into active enzymes will also be attempted. Here we report successful recloning and over-expression of recombinant, short-type Pr-MnP3 in E. coli, the recovery of active rPr-MnP3 after in vitro refolding and preliminary characterization of the recombinant enzyme.
2. MATERIALS AND METHODS
2.1. Materials
DNA manipulations, growth media and buffers were carried out according to the method of [45]. All chemicals used in this investigation were of reagent grade and obtained from Fisher or Sigma-Aldrich chemicals Co as pure commercial products and used without further purification. All restriction enzymes and their appropriate buffers were supplied by NEB (UK) Ltd. All Spectroscopic measurements were carried out using a ShimazduUV-2401 spectrophotometer. The synthesis of oligonucleotide primers and DNA sequencing was performed by MWG-biotech, UK. The MnP3 cDNA [9] cloned into the vector (pCR2.1TOPO) was generously provided by Dr. Taina Lundell, Department of Food and Environmental Sciences, Division of Microbiology, University of Helsinki, Finland. The GeneBank accession number for MnP3 cDNA/pCR2.1.TOPO is AJ566200. The E. coli expression vector pFLAG1 was obtained from International Biotechnologies Inc.
2.2. Primer Design
P. radiata MnP3 full length coding region cDNA was previously cloned in E. coli as an insert in pCR2.1TOPO vector [20]. Two primers, for the mature N-terminal and C-terminal ends of Pr-MnP3 were designed based on previous information on Pr-MnP3 gene [20] for amplifying and cloning the coding sequence into the E. coli expression vector (pFLAG1). Oligonucleotide Pr-MnP3-Nt (5’ GAATTC CAT ATG TTA ACT GTG GCT TGC CCA GAT GGT GTG AAC ACT GC 3’) corresponding to the first ten residues of the mature sequence was preceded by a sequence containing EcoRI and an NdeI restriction sites. The design of the N-terminal primer involved the removal of signal sequence, the introduction of three amino acids, methionine, leucine and threonine (MLT) encoding codons and optimisation of codon usage and GC content for E. coli. Oligonucleotide Pr-MnP3-Ct (5’- GAATTC GGATCC TTA CGA CGG GGG GAC AGG CGC AAC AGA-3’) was reverse and complementary to the last 8 codons of the MnP3 cDNA with additional BamHI and EcoRI restriction sites following the stop codon.
2.3. Generation of the Engineered Phlebia radiata MnP3 Gene by the PCR
The engineered Pr-MnP3 was amplified by the PCR technique using Pfu DNA polymerase (Stratagene), which has 3’ - 5’ proof-reading activity in a programmable thermal cycler (Perkin Elmer GeneAmp PCR System 9600). 5 ng of DNA template (P. radiata MnP3), 1 µM each of oligonucleotides (MnP3-Nt and MnP3-Ct) 10 µM each dNTP, 1 µl of 10× Pfu polymerase buffer and 2.5 units of Pfu DNA polymerase in a final volume of 50 µl were in the reaction mixture. Initial denaturation was carried out at 95˚C for 10 minutes. This was followed by 25 cycles: each comprising of denaturation (1 minute at 95˚C), annealing (1 minute at 60˚C), and extension (5 minutes at 72˚C). The PCR product was electrophoretically analyzed using 1% agarose gel.
2.4. Cloning of the Engineered Phlebia radiata MnP3 Gene into the E. coli Expression Vector pFLAG1
The engineered Pr-MnP3 was first cloned into a commercially available pCR 2.1TOPO TA cloning vector obtained from Invitrogen, UK. It was the cloned into E. coli expression vector (pFLAG1). The cloning was facilitated by treating the PCR product first with Taq polymerase and dNTPs. The Taq polymerase has a nontemplate dependent terminal transferase activity that adds a single deoxyadenosine (A) termini overhang to 3’ ends of PCR fragments. 1 µl of Taq polymerase and 5 µl of dNTP were added to the PCR product and the mixture was incubated in a thermal cycler for 30 minutes at 72˚C but without cycling. This was to ensure the presence of 3’A-overhangs at the ends of PCR fragments, since pCR2.1TOPO TA cloning vector is supplied in a linearised form with a single 3’ deoxythymidine (T) residue overhang. A one step cloning strategy for the direct insertion of Taq treated PCR products into a pCR2.1 TOPO TA plasmid vector was carried out and the presence of A-overhang at the 3’ends of the PCR inserts facilitated efficient cloning. The vector (topoisomerase I activated) also contain ampicillin and kanamycin resistance marker. The cloned pCR2.1-MnP3 was further transformed into chemically competent E. coli (TOP10). The transformation processes involved the addition of 2 µl of the reaction solution to chemically competent cells and incubate on ice for 15 minutes. The “heat shocked” process lasted for 30 seconds at 42˚C before it was placed on ice for 2 minutes to recover. Thereafter, 250 µl of LuriaBertani (LB) medium was added and incubated at 37˚C for 1 hour with shaking at 170 rpm. The solution was centrifuged at 6000 rpm for 2 minutes and the supernatant were discarded leaving approximately 100 µl in the tube. The transformed cells were grown on L-agar plate containing suitable antibiotic (kanamycin 25 µg/µl), 100 µl of 20 µg/ml of X-gal and 1 ml of 0.2 mg/ml of IPTG. The pCR2.1 contains the TA cloning site within the lacZ gene, under the control of a lac promoter, which allows the selection of clones that contain insert by “blue/white” colony. Small-scale plasmid DNA preparation (Miniprep) using QIAprep Spin Miniprep Kit protocol (QIAGEN) was used to purify plasmid DNA from E. coli cultures. The resultant putative pCR2.1-MnP3 DNA colonies were diagnostically screened using EcoRI restriction enzyme and the digest evaluated by 1% agarose gel electrophoresis. Positive clones were identified and cultured in a medium overnight culture containing L-broth with 25 mg/ml of kanamycin at 37˚C with the rpm 170 DNA was extracted using the Promega Wizard® Plus Midipreps DNA purification system kit (Promega). The correct DNA sequence and reading frame was confirmed by automatic sequencing by MWG-Biotech using with universal and reverse primers. The cloned pCR2.1- MnP3 DNA was purified using Geneclean BIO 101 kit, and the DNA was digested with NdeI and BamHI and recloned into the commercially available ampicillin-resistant E. coli expression vector (pFLAG1) International Biotechnologies Inc., UK. The resulting construct, pFL AG1-MnP3 (Figure 1) was transformed into E. coli (W3110).
2.5. Site-Directed Mutagenesis
The mutations were introduced by PCR based method (Whole Plasmid Amplification Method (WPAM)) [46] using pFLAG1-MnP3 as template and two primer designed strategies. E40H and E44H mutations were introduced by replacing Glu 40 and Glu 44 with Histidine (His) residue and the double mutant (E40H/E44H) by substituting Glu 40/Glu 44 with Histidine at both positions. For each mutant, both a mutagenic and a reverse primer were designed complementary to opposite strands of the same DNA region. Oligonucleotide sequences were as follows: E40H mutagenic primer, 5’-CAC CTC GTG GCC ACA CTC GCC GCC-3’; E40H reverse primer, 5’-CAC CTC CTC GCC ACA CTC GCC-3’; E44H mutagenic primer, 5’-CAC CAC TCT CTG CGC CTC ACG TTC-3’, E44H reverse primer, 5’-CAC CTC CTC GCC ACA CTC GCC-3’. The mutations to the wild-
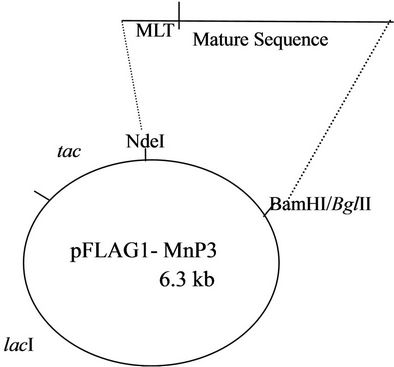
Figure 1. An engineered Phlebia radiata MnP3 gene, including the three residues (MLT) added to the beginning of the gene was cloned into a multicopy expression plasmid (pFLAG1) under tac promoter control, to yield the vector pFLAG1-MnP3.
type are under-lined. The double mutant, E40H/E44H was obtained as described for the single ones and mutagenic and reverse primers were those of E40H and E44H. It is important to note that the ApaL I restriction site (GTG CAC) is present in pFLAG1-MnP3. The gene was further transformed in E. coli (DH5α), and digested with ApaL I to select the correct integrity of the ligation site.
Back-to-back primers were designed to introduce sitespecific D186N and D186H mutations with incorporation of silent mutations creation of PvuII restriction site (CAG CTG) for identification of the mutant clones. Oligonucleotide sequences were as follows: D186N mutagenic primer, 5’-CTG CCA ATC ACG TCG ACC CAT CGA TCC-3’; D186N reverse primer, 5’-CTG CGA TCG TGT GCG ACG CGA G-3’; D186H mutagenic primer, 5’- CTG CCC ATC ACG TCG ACC CAT CGA TCC-3’; D186H reverse primer, 5’-CTG CGA TCG TGT GCG ACG CGA G-3’. The mutations to the wild-type are underlined. For each mutant, both primers coincided exactly back-to-back on opposite strands of the template DNA (pFLAG1-MnP3). After amplification, a linear DNA fragment identical in size to linearized pFLAG1-MnP3 was obtained and purified using Dpn I endonuclease to cleav off the methylated wild-type template. The mutant DNA was phosphorylated using T4 polynucleotide kinase, ligated using T4 DNA ligase and transformed into E. coli (DH5α). Positive colonies were selected on the basis of digestion of their plasmidic DNA by either ApaLI or PvuII, thus completely excluding wild-type plasmids and mutant plasmids with deletions due to spurious exonuclease activity of polymerase. After complete automated sequencing, one of the positive clones for each mutant was used to transform E. coli (W3110 strain).
All PCR reactions were conducted in a Perkin-Elmer GeneAmp PCR System 9600 using 5 ng of template DNA, 1 mM each dNTP, 10 µM of mutagenic and reference primer, 10 units of Pfu polymerase and the Pfu reaction buffer (Promega). The reaction conditions were as follows: (i) a “hot start” of 95˚C for 10 minutes and 25 cycles of denaturation (95˚C for 1 minute), annealing (50˚C for 1 minute), extension (72˚C for 16 minutes) and finally at 4˚C forever (∞).
2.6. Expression of the Engineered Pr-MnP3 Genes
The small-scale expression of engineered Pr-MnP3 gene was carried out by overnight culture of pFLAG1-MnP3 in Luria-Bertani broth which was inoculated into 20 ml fresh cultures in a 100 ml conical flask. Approximately after 3 hours growth (A500 ≈ 0.8), expression was induced for a further 3 hours with 1 mM IPTG. Bacteria from 3 ml of each culture were harvested at 6000 rpm for 2 min in a microcentrifuge and resuspended in 500 µl of lysis buffer (20 mM Tris-HCl (pH 8.0), 1 mM EDTA, 1 mM dithiothreitol (DTT) and 6 M urea). The resuspended cells were sonicated using MSE 150 W Ultrasonic Disintegrator MK2 (Crawley, UK) on ice for 30 seconds, centrifuged for 15 min at 13,000 rpm and the supernatant was discarded. The protein containing inclusion body pellets were resuspended in 500 µl of the lysis buffer and resonicated. An equal volume of the lysis buffer supplemented with 3% Triton X-100 was added to the resonicated protein solution and kept at room temperature for 10 min before centrifugation for 15 min at 13,000 rpm. The resulting pellets were resuspended by vigorous vortexing in 30 µl of 50 mM Tris-HCl buffer (pH 8.0); containing 1 mM EDTA, 30 mM DTT, 10 M urea and centrifuged at 13,000 rpm for 10 minutes. The presumed rPr-MnP3 containing supernatant was run on an SDS-PAGE was used to identify the rPr-MnP3 protein in the supernatant (Figure 3).
The small-scale expression reported here provided evidence of successful expression of rPr-MnP3 which culminated into large-scale production of rPr-MnP3 in E. coli (W3110) cells. In the large-scale expression, the E. coli (W3110) cells containing pFLAG1-MnP3 were grown in 2 litres flasks containing 500 ml of Terrific Broth (TB) and 100 µg/ml ampicillin. After 3 hours growth at 37˚C (OD500nm ~ 1) the expression was induced with 1 mM IPTG and cultivated for 3.5 hours. The cells were harvested by centrifugation at 4000 rpm using Beckman Centrifuge (Model J-6B).
2.7. Isolation and Solubilisation of Unfolded Inclusion Bodies of the rPr-MnP3 Protein
The rPr-MnP3 pellets were resuspended in lysis buffer (50 mM Tris-HCl (pH 8.0) containing 1 mM EDTA, 30 mM DTT and 10 M urea and lysozyme was added to a final concentration of 2 mg/ml. After 45 min incubation at room temperature, the cells were further disrupted by sonication for 2 min and Centrifuged at 15,000 rpm for 30 min to obtain inclusion bodies containing rPr-MnP3 protein. The inclusion body protein pellet was resuspended by homogenisation in wash buffer (20 mM TrisHCl, pH 8.0, 1 mM EDTA and 5 mM DTT). Membrane components and proteases were removed achieved by washing the resuspended pellet with the wash solution containing 3% Triton X-100 and recentrifuged at 15,000 rpm for 30 minutes. The process of resuspension and washing was repeated thrice. For further experiments, rPr-MnP3 protein was solubilised using 50 mM Tris-HCl buffer (pH 8.0) containing 1mM EDTA, 1 mM DTT and 6 M urea for 15 min before centrifugation at 15,000 rpm for 30 min to remove remaining insoluble materials.
2.8. Folding, Activation and Purification of rPr-MnP3
The protein concentration of the unfolded rPr-MnP3 protein were estimated using the dye-binding method (BioRad Laboratories, UK) using BSA as standard for total protein. Small-scale refolding recombinant P. radiata MnP3 into active protein was carried out according to methods for refolding of plant peroxidases [30,31,33]. Finally, large-scale refolding of recombinant P. radiata MnP3 enzymes was performed using the optimal conditions reported [33]. The refolding medium consisted of the following: 50 mM Tris-HCl, pH 9.5, 5 mM CaCl2, 0.5 mM GSSG, 0.1 mM DTT, 0.15 M urea, 20 µM haemin and 200 µg/ml protein. After the addition of protein solution into the folding medium with stirring, the folding solution was divided into 400 ml aliquots in 1000 ml beakers and stored overnight in the dark at room temperature. The folding mixture was concentrated 20-fold using Amicon spiral-wound concentrator. It was further concentrated to approximately one-sixth of its initial volume using an Amicon stirred ultrafiltration cell with a PM-10 membrane. The resultant enzyme solution was extensively dialysed against 20 mM sodium acetate, pH 4.3, containing 1 mM CaCl2, in the cold room overnight before centrifuged at 15,000 rpm (Beckman J2-21) for 30 minutes to remove aggregates. The solution was further dialysed against 10 mM sodium succinate, pH 6.0, and 1 mM CaCl2 solution in the cold room overnight and recentrifuged at 15,000 rpm for 30 minutes. The protein solution in 10 mM sodium succinate, pH 6.0, containing 1 mM CaCl2 was syringe-filtered before loaded into a 50 ml Mono-Q anion exchange column (Pharmacia). The active rPr-MnP3 enzyme was eluted with a linear gradient of NaCl from 0 to 0.5 M with rPr-MnP3 appearing as a sharp peak at approximately 300 mM NaCl. The fractions that contained MnP3 with high activity and Soret peak at 408 nm were collected and gel-filtered into 10 mM sodium succinate, pH 6.0 buffer containing 0.2 mM CaCl2 using a PD10 desalting column (Pharmacia). The purified rPr-MnP3 was frozen in liquid nitrogen and stored in a freezer at –80˚C.
2.9. Characterisation of MnP3 Enzyme
2.9.1. Mass Estimation
The molecular mass of the recombinant Pr-MnP3 enzymes was determined by SDS-PAGE on 12% polyacrylamide gels, using a Bio-Rad Mini-protean II electrophoresis cell with marker proteins (Sigma). The gels were stained with Coomasie blue R-250. The molecular mass was also accurately determined using MALDI-TOF MS (Mass Spectrometry Centre, University of Sussex).
2.9.2. Spectroscopic Measurements
Resting-state UV-visible spectra of rPr-MnP3 enzymes in 10 mM sodium succinate, pH 6.0 were recorded at 25˚C using a Shimadzu UV-2401 PC spectrophotometer (250 - 750 nm). The concentration of wild-type rPr-MnP3 was calculated from the absorption at 408 nm of the resting state spectrum using an extinction coefficient of 168 mM–1·cm–1 [47]. The Reinheitszahl number (RZ), which gives a convenient measure of purity was also estimated using the ratio of the absorbance at the Soret peak and at 280 nm (ASoret/A280).
2.9.3. Measurement of Extinction Coefficient
The extinction coefficient of the Soret peak for rPr-MnP3 enzymes was determined by the pyridine haemochrome method and calculated using the published extinction coefficient for the reduced-oxidised haem (A555 – A542) = 20.7 mM–1·cm–1 [47].
3. RESULTS AND DISCUSSION
Two primers, 5’ and 3’ to the Pr-MnP3 cDNA readingframe were made to engineer the Nand the C-termini of the MnP3 protein and to incorporate restriction sites for in-frame cloning. The E. coli initiation codon AUG was introduced in frame with the first codon of the mature sequence via an NdeI restriction site included at the N-terminus. These primers allowed the amplification of a full length cDNA fragment (Figure 1), encoding the mature Pr-MnP3 protein sequence without the preprosequence, and also adding to the 5’ end codons for three additional amino acid residues (M, L, and T) to enhance expression in E. coli. After confirmation of the correct DNA sequence present in pCR2.1TOPO (Figure 2) by automatic sequencing, the engineered cDNA was recloned at the NdeI and BglII restriction sites of pFLAG1 and the resulting pFLAG1-MnP3 construct and that of the mutants were transformed into E. coli (W3110 strain).
E40H, E44H, E40H/E44H, D186H and D186N mutants were generated from the wild-type rPr-MnP3 gene.
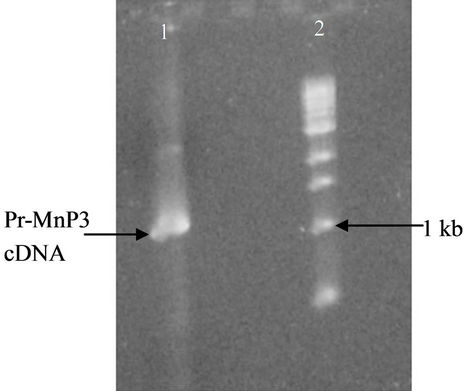
Figure 2. 1 % agarose gel of PCR product showing engineered rPr-MnP3 (lane 1) and DNA molecular size marker (lane 2).
The numbering 40, 44, and 186 of the mutant genes is for our modified wild-type rPr-MnP3 protein with 3 additional wild-type rPr-MnP3 protein with 3 additional amino acid residues (M, L and T) at the N-terminus to enhance protein expression However, the equivalent database number for Phlebia radiata wild-type MnP3 are E37, E41 and D183 and the sequence alignment of wildtype Pr-MnP3 with Phanerochaete chrysosporium MnP identified E37, E41 and D183 as the ligands for Mn (II) binding site of the MnP3 enzyme.
3.1. Expression in E. coli W3110 and Extraction of the rPr-MnP3 from Inclusion Bodies
The use of PCR to produce an engineered gene for the over expression of rPr-MnP3 isoenzyme in E. coli W3110 was very successful. The engineered rPr-MnP3 gene contained the gene sequence encoding the mature protein sequence. The engineering involved the removal of the pro-sequence and the addition of 3 amino acid residues (MLT) at the beginning of the 5’ end. The addition of the MLT was to enhance in-frame cloning into expression vector (pFLAG1). The gene was cloned into the commercially available E. coli expression vector, pFLAG1 under the control of the tac promoter giving rise to pFLAG1-MnP3 (Figure 1).
Expression of MnP3 protein from the engineered gene in E. coli W3110 was found to be inducible by the addition of IPTG. The MnP3 was sequestered into insoluble inclusion bodies like other recombinant peroxidase previously expressed in E. coli [30-32,35,42]. The inclusion bodies (approximately 80% rPr-MnP3 protein) were partially purified by use of 1% Triton X-100. To obtain active enzyme, the inclusion body protein was then solubilised using a large amount of 6 M urea and then refolded. The size of the rPr-MnP3 proteins using SDSPAGE was found to be approximately 36 kDa as predicted from the engineered gene sequence (Figures 3 and 4).
3.2. Refolding and Purification of Active Recombinant Pr-MnP3 Enzyme
In the refolding of the recombinant P. radiata MnP3 in vitro, we tried methods reported for other recombinant plant and fungal haem-containing peroxidases, such as HRP [30], LiP H8 from P. chrysosporium [32] and Versatile Peroxidase (VP) from Pleurotus eryngii [34]. However, the VP refolding conditions (50 mM Tris-HCl, pH 9.5, 5 mM CaCl2, 0.5 mM GSSG, 0.1 mM DTT, 0.15 M urea, 20 µM haemin and 0.1 mg/ml protein at pH 9.5) was adopted in this work since it gave a significant level of activity with ABTS from refolding mixture. Following refolding of the protein, the resultant solution was concentrated and dialysed as described in the methodology section before a final purification step, using ion ex

Figure 3. SDS-PAGE analysis of insoluble inclusion bodies after small-scale expression of rPr-MnP3. Inclusion body protein pellets were solubilized in 6 M urea and loaded on a 12 % resolving SDS-PAGE protein gel. Lane 1 is rHRP (Control). Lanes 2 and 3 induced MnP3 samples. Lane 4 is the protein molecular weight marker (full-range Rainbow, Biorad laboratories). Gel was stained with Comassie Blue. As predicted from the engineered gene sequence, the SDS-PAGE gel showed a clearly visible band of expressed rPr-MnP3 with a molecular mass of about 36 kDa.
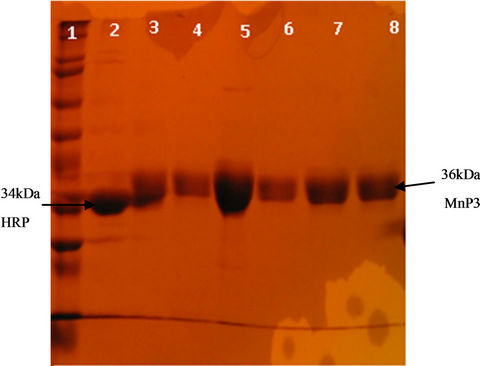
Figure 4. SDS-PAGE of P. radiata purified wild-type MnP3 protein and mutant variants isolated from inclusion bodies. Protein extracts were loaded on a 12% resolving SDS-PAGE protein gel. Lane 1 Molecular weight marker, lane 2 HRP, lane 3 D186H, lane 4 D186N, lane 5 wild-type, lane 6 E40H/E44H, lane 7 E40H and lane 8 E44H. Bands were visualized using Comassie Blue staining. Note that the wild-type MnP3 and HRP proteins were from purified samples.
change on a FPLC system (Parmacia). A gradient of over 30 ml of 0 - 1 M NaCl was run through a Mono Q anion exchange column, and a large protein peak was eluted at approximately 300 mM NaCl. This peak was coloured and in assays showed high level of oxidation of Mn2+. Therefore, the active fractions were pooled and filtered using gel filtration method into 10 mM Na succinate, pH 6.0 containing 0.2 mM CaCl2. Figure 5 shows a typical
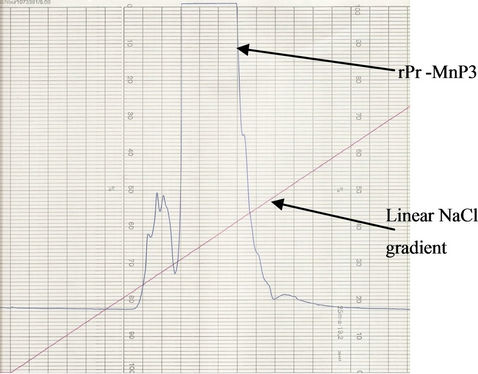
Figure 5. Typical chromatogram for purification of P. radiata recombinant MnP3. Active rPr-MnP3 enzyme was purified using the FPLC with a Mono-Q anion exchange column in 10 mM Na succinate buffer, pH 6.0, 1 mM CaCl2 with a linear NaCl gradient of 0 - 1 M as indicated by (—). The rPr-MnP3 enzyme eluted, as indicated by A280 (—) peak, at approximately 300 mM NaCl.
elution profile obtained for P. radiata recombinant MnP3 enzyme.
Approximately 7 mg of active rPr-MnP3 enzymes were purified from a 5 litre E. coli expression, after refolding and purification. The refolding efficiency was 6% - 7 % for the wild-type and each of the mutants, which is similar to that of rHRP and rCiP after refolding [30].
3.3. Enzyme Characterizations
3.3.1. Spectroscopic Characteristics
The UV/visible spectrum (Figure 6) of the engineered wild-type rPr-MnP3 contains the Soret band maximum at 408 nm, and additional lower bands at 507 and 641 nm.
These spectral characteristics of rPr-MnP3 are very similar to those of P. radiata MnP2 with 407 nm Soret maximum and 508 and 640 nm absorbance bands [7], and P. chrysosporium MnPH4, with 406 nm Soret maximum and 502 and 632 nm lower absorbance bands, respectively [17]. The spectral characteristics of rPr-MnP3 as shown in Figure 6 is typical of a substantially highspin haem protein, probably a little more 6-coordinate like LiP, but clearly distinct from a 5-coordinate classical high-spin peroxidases such as HRP.
The spectral characteristics of the wild-type rPr-MnP3 and Mn site mutant enzymes as shown in Table 1 and Figure 7 indicate that these enzymes are very similar except E40H. This means that mutations at the Mn-binding site appear to cause minimal distortion to the haem environment. A small but significant effect in the visible region of the E40H was observed relative to the other rPr-MnP3 enzymes. He CT I appeared at 629 nm and the
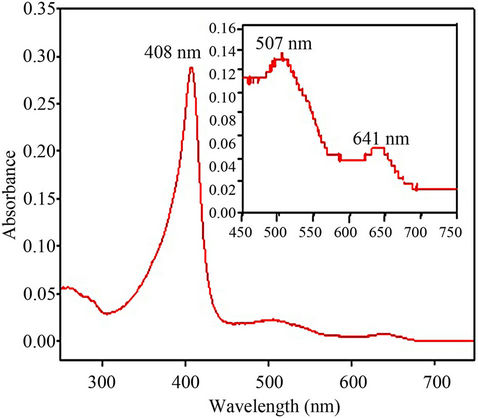
Figure 6. Resting state UV/Visible absorption spectrum for purified wild-type recombinant P. radiata MnP3. The spectrum was recorded in 10 mM sodium succinate, pH 6.0 at 25˚C. The Soret maximum is at 408 nm, while the insert is the magnified region of 450 - 750 nm showing the charge transfer bands at 641 and 507 nm.
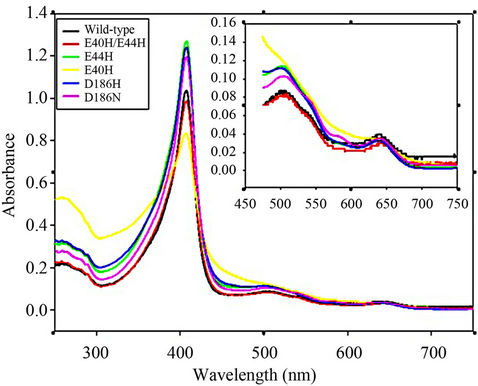
Figure 7. Overlay of the resting state UV/Visible absorption spectra for recombinant P.radiata wild-type MnP3 and mutant variants. The spectra were recorded in 10 mM sodium succinate, pH 6.0. (―) wild-type, (―) E40H/E44H, (―) E44H, (―) E40H, (―) D186H and (―) D186N. The insert to the figure on the upper right corner is a magnification of the 450 to 750 nm region.
Soret at 408 nm, while CT II was not observable by the steady state method. However, significant light scattering was evident only in the E40H spectrum, indicating some aggregation of the protein. All attempts to remove the scattering by spinning the enzymes at 13,000 rpm for 15 minutes and filtering through a 0.22 nm filter did not alter the spectrum. It is believed that the aggregation is a peculiar property of E40H mutant as it was not observed in any other mutant generated in this study.
The wild-type and four rPr-MnP3 mutant (E44H, E40H/E44H, D186H and D186N) enzymes in this study have RZ values greater than 4 (Table 1). This is indicative of high purity enzymes suitable for kinetic studies. However, the RZ of 2.1 reported for the E40H mutant is lower than that of others, likely due to aggregation. The RZ is thus in principle a measure of haem content relative to the aromatic amino acid content of a sample. In the case of HRP, a 5-coordinate haem protein, high purity samples have RZ values in the range 3.0 - 3.5 [49-51]. RZ values of 5.1 and 5.9 have been reported for 6-coordinate haem enzyme like LiP and MnP respectively [52].
The extinction coefficient of the Soret peak, which gives a measure of the absorption strength of the haem in the context of the protein environment, was determined by the pyridine hemochrome method [53]. In this study, the absorption spectrum of reduced pyridine hemochrome complex of rPr-MnP3 exhibited maxima at 556 nm (α),
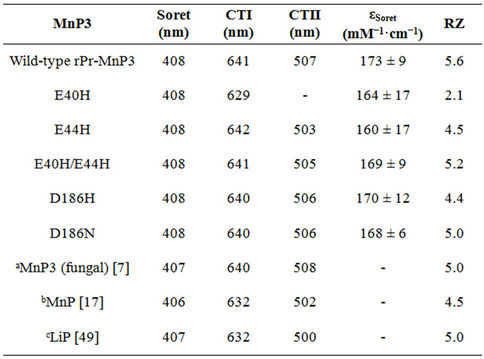
Table 1. Summary of the spectroscopic characteristics of resting state P. radiata wild-type and mutant MnP3 enzymes.
522 nm (β) and 419 nm (γ). Also the recombinant PrMnP3 Soret extinction coefficient (mM–1·cm–1) values for wild-type (173 ± 9) and that of the mutants, E40H (164 ± 17), E44H (160 ± 17), E40H/E44H (169 ± 9), D186H (170 ± 12) and D186N (168 ± 6) are reported in this investigation.
The observed values are within the range obtained for other fungal secreted class II peroxidases with an established six-coordinate haem iron [48] and pyridine haemochromes of protoporphyrin IX [53].
3.3.2. Mass Characterization
SDS-PAGE and MALDI-TOF MS methods were employed in the mass characterisation of the enzymes and it was observed that both the wild-type and mutant rPrMnP3 enzymes had the molecular mass of approximately 36 kDa. The MALDI-TOF data for the engineered wild-type and the mutant rPr-MnP3 enzymes are presented in Figures 8(a)-(f). The additional peaks are due to the internal standards, which are necessary for accurate MALDI-TOF mass determinations of protein with molecular mass above 20 kDa [54]. The MALDI-TOF mass spectral peak and SDS-PAGE band of rPr-MnP3 are at 36.065 kDa, compared to a calculated protein mass of 35.703 kDa (DNAstar software). This result indicates a monomeric enzyme, similar to those described for other fungal enzymes such as MnPs, LiPs and VPs [55- 57].
4. CONCLUSION
Wild-type MnP3 gene from P. radiata has been engineered for high level E. coli expression and Mn (II) binding site mutants (E40H, E44H, E40H-E44H, D186H and D186N rPr-MnP3) generated by PCR-based method (WPAM). Both the wild-type and mutant rPr-MnP3 genes were successfully expressed in E. coli and the enzymes
 (a)
(a)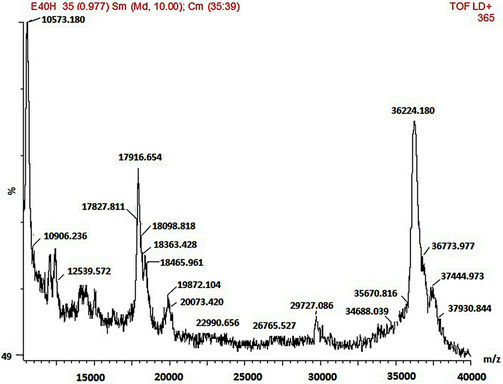 (b)
(b)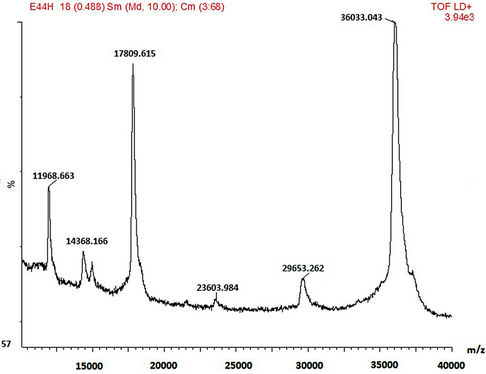 (c)
(c)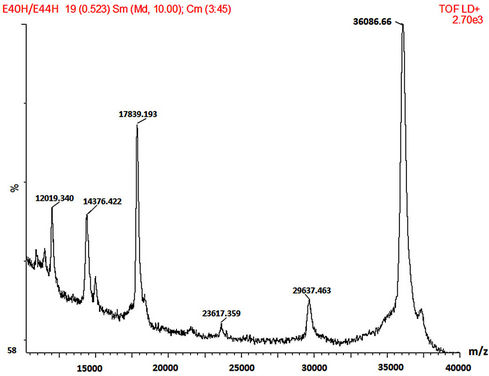 (d)
(d)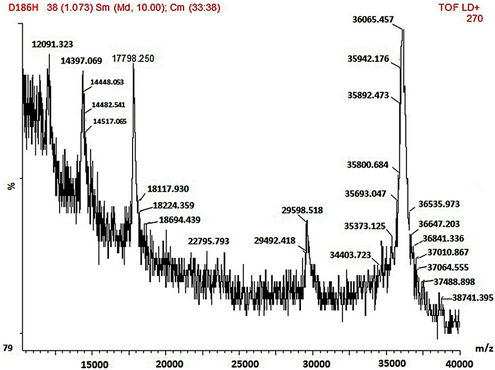 (e)
(e)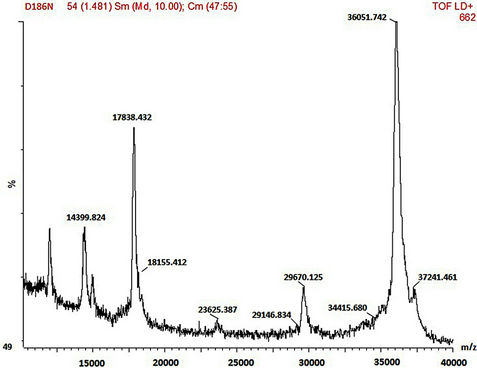 (f)
(f)
Figure 8. (a)-(f): MALDI-TOF mass spectrum of the engineered wild-type and mutants MnP3 from P. radiata. A = wild type MnP3; B = E40H; C = E44H; D = E40H/E44H; E = D186H; F = D186N.
refolded and purified, for the first time. The methods used for refolding of rPr-MnP3 enzymes is an adaptation of one previously used for the salvage of recombinant versatile peroxidase from E. coli inclusion bodies [34]. Heterological expression and reconstitution of recombinant rPrMnP3 enzymes from Phlebia radiata yielded a remarkable success (6% - 7% recovery of active rPr-MnP3). The yield attained under the rPr-MnP3 in vitro folding conditions established in the present study was similar to that obtained for VPL2 (up to 7% folding yield) [33], but higher than that reported for other recombinant secretory haem-containing peroxidases belonging to Classes II and III [30-32]. The molecular mass of the rPr-MnP3 enzymes in this study was determined to be 36 kDa approximately and the spectroscopic characterization of the P. radiata enzymes revealed a typical high spin haem 6-coordinate peroxidases. This achievement will remain invaluable in a variety of mechanistic studies and also greatly facilitate future studies on structure-function relationships of rPrMnP3 enzymes. Work on full characterization of the engineered rPr-MnP3 enzyme and its site-directed mutants are ongoing in our laboratories.
5. Acknowledgements
We would like to thank Professor Andrew Smith for providing the Laboratory space and technical assistance during this study. Dr. Wendy Doyle is gratefully acknowledged for her assistance during the laboratory work. We are also grateful to Dr. Taina Lundell for providing the cDNA; and to The Akwa Ibom State University, Akwa Ibom State, Nigeria, for sponsoring this research.
REFERENCES
- Hatakka, A.I. and Uusi-Rauva, A.K. (1983) Degradation of it labeled poplar wood lignin by selected white-rot fungi. European Journal of Applied Microbiology Biotechnology, 17, 235-242. doi:10.1007/BF00510422
- Lundell, T., Leonowicz, A., Rogalski, J. and Hatakka, A. (1990) Formation and action of lignin modifying enzymes in cultures of Phlebia radiata supplemented with veratric acid. Applied Environmental Microbiology, 56, 2623-2629.
- Vares, T., Kalsi, M. and Hatakka, A. (1995) Lignin peroxidases, manganese peroxidases, and other ligninolytic enzymes produced by Phlebia radiata during solid-state fermentation of wheat-straw. Applied Environmental Microbiology, 61, 3515-3520.
- Hatakka, A., Lundell, T., Hofrichter, M. and Maijala, P. (2002) Manganese peroxidase and its role in the degradation of wood lignin. In: Mansfield, S.D. and Saddler J.N., Eds., Applications of Enzymes to Lignocellulosics, American Chemical Society Symposium Series, New York, 7-15.
- Niemenmaa, O., Uusi-Rauva, A.K. and Hatakka, A. (2006) Wood stimulates the demethoxylation of [O14CH3]-labeled lignin model compounds by the white-rot fungi Phanerochaete chrysosporium and Phlebia radiata. Archives of Microbiology, 185, 307-315. doi:10.1007/s00203-006-0097-5
- Lundell, T. (1993) Ligninolytic system of the white-rot fungus Phlebia radiata: Lignin model compound studies. Ph.D. Thesis, Department of Applied Chemistry and Microbiology, University of Helsinki, Helsinki.
- Karhunen, E., Kantelinen, A. and Niku-Paavola, M. (1990) Mn-dependent peroxidase from the lignin-degrading white rot fungus Phlebia radiata. Archives of Biochemistry and Biophysics, 279, 25-31. doi:10.1016/0003-9861(90)90458-B
- Moilanen, A.M., Lundell, T., Vares, T. and Hatakka, A. (1996) Manganese and Malonate are individual regulators for the production of lignin and manganese isozymes and in the degradation of lignin by Phlebia radiata. Applied Microbiology and Biotechnology, 45, 792-799. doi:10.1007/s002530050764
- Hilden, K.S., Makela, M.R., Hakala, T.K., Hatakka, A. and Lundell, T. (2006) Expression on wood, molecular cloning and characterization of three lignin peroxidase (LiP) encoding genes of the white rot fungus Phlebia radiata. Current Genetics, 49, 97-105. doi:10.1007/s00294-005-0045-y
- Lundell, T. K., Makela, M.R. and Hilden, K. (2010) Lignin-modifying enzymes in filamentous basidiomycetes— ecological, functional and phylogenetic review. Journal of Basic Microbiology, 50, 5-20. doi:10.1002/jobm.200900338
- Hofrichter, M., Ullrich, R., Pecyna, M.J., Liers, C. and Lundell, T. (2010) New and classic families of secreted fungal heme peroxidases. Applied Microbiology and Biotechnology, 87, 871-897. doi:10.1007/s00253-010-2633-0
- Glenn, J.K. and Gold, M.H. (1985) Purification and Characterization of an extracellular Mn (II)-dependent peroxidase from the lignin—degrading basidiomycete, Phanerochaete chrysosporium. Archives of Biochemistry and Biophysics, 242, 329-341. doi:10.1016/0003-9861(85)90217-6
- Paszczynski, A., Huynh, V.B. and Crawford, R. (1985) Enzymatic activites of an extracellular, manganese-dependent peroxidase from Phanerochaete chrysosporium. FEMS Microbiology Letters, 29, 37-41. doi:10.1111/j.1574-6968.1985.tb00831.x
- Hofrichter, M. (2002) Review: Lignin conversion by manganese peroxidase (MnP). Enzyme Microbial Technology, 30, 454-466. doi:10.1016/S0141-0229(01)00528-2
- Kirk, T.K., Croan, S.D., Tien, M., Murtagh, K.E. and Farrell, R.L. (1986) Production of multiple ligninases by Phanerochaete chrysosporium: Effect of selected growth conditions and use of a mutant strain. Enzyme Microbial Technology, 8, 27-32. doi:10.1016/0141-0229(86)90006-2
- Pease, E.A., Andrawis, A. and Tien, M. (1989) Manganese-dependent peroxidase from Phanerochaete chrysosporium. Primary structure deduced from complementary DNA sequence. Journal of Biological Chemistry, 264, 13531-13535.
- Mayfield, M.B., Kishi, K., Alic, M. and Gold, M.H. (1994) Homologous expression of recombinant manganese peroxidase in Phanerochaete chrysosporium. Applied Environmental Microbiology, 60, 4303-4309.
- Alic, M., Akileswaran, L. and Gold, M.H. (1997) Characterization of the gene encoding manganese peroxidase isozyme 3 from Phanerochaete chrysosporium. Biochimica et Biophysica Acta, 1338, 1-7. doi:10.1016/S0167-4838(96)00235-X
- Martinez, D., et al. (2004) Genome sequence of the lignocellulose degrading fungus Phanerochaete chrysosporium strain RP78. Nature Biotechnology, 22, 695-700. doi:10.1038/nbt967
- Hilden, K., Martinez, A.T., Hatakka, A. and Lundell, T. (2005) The two manganese peroxidases pr-Pr-MnP2 and Pr-MnP3 of Phlebia radiata, a lignin-degrading basidiomycete, are phylogenetically and structurally divergent. Fungal Genetics and Biology, 42, 403-419. doi:10.1016/j.fgb.2005.01.008
- Martinez, A.T. (2002) Molecular biology and structurefunction of lignin-degrading heme peroxidases. Enzyme Microbial Technology, 30, 425-444. doi:10.1016/S0141-0229(01)00521-X
- Forrester, I.T., Grabski, A.C., Mishra, C., Kelly, B.D., Striekl, W.N., Leatham, G.E. and Burgess, R.R. (1990) Characteristics and N-terminal amino acid sequence of a manganese peroxidase purified from Lentinula edodes culture grown on a commercial wood substrate. Applied Microbiology and Biotechnology, 33, 359-365. doi:10.1007/BF00164536
- Glenn, J.K., Akileswaran, L. and Gold, M.H. (1986) Mn(II) oxidation is the principal function of extracellular Mn-peroxidase from Phanerochaete chrysosporium. Archives Biochemistry and Biophysics, 251, 688-696. doi:10.1016/0003-9861(86)90378-4
- Sundaramoorthy, M., Terner, J. and Poulos, T.L. (1995) The crystal structure of chloroperoxidase: A heme peroxidase-cytochrome P450 functional hybrid. Structure, 3, 1367-1377. doi:10.1016/S0969-2126(01)00274-X
- Perez, J. and Jeffries, T.W. (1992) Roles of manganese and organic acid chelators in regulating lignin degradation and biosynthesis of Phanerochaete chrysosporium. Applied Environmental Microbiology, 58, 2402-2409.
- Niku-Paavola, M.L., Karhunen, E., Salola, P. and Raunio, V. (1988) Ligninolytic enzymes of the white-rot fungus Phlebia radiata. Biochemistry Journal, 254, 877-883.
- Lundell, T., Wever, R., Floris, R., Harvey, P., Hatakka, A., Brunow, G. and Schoemaker, H. (1993) Lignin peroxidase L3 from Phlebia radiata. Pre-steady-state and steady-state studies with veratryl alcohol and a non-phenolic lignin model compound 1-(3,4-dimethoxyphenyl)- 2-(2-methoxyphenoxy) propane-1, 3-diol. European Journal of Biochemistry, 211, 391-402. doi:10.1111/j.1432-1033.1993.tb17562.x
- Orth, A.B., Royse, D.J. and Tien, M. (1993) Ubiquity of lignin-degrading peroxidases among various wood-degrading fungi. Applied Environmental Microbiology, 59, 4017-4023.
- Lundell, T. and Hatakka, A. (1994) Participation of Mn (II) in the catalysis of laccase, manganese peroxidase and lignin peroxidase from Phlebia radiata. FEBS Letter, 348, 291-296. doi:10.1016/0014-5793(94)00627-X
- Fishel, L.A., Villafranca, J.E., Maurao, J.M. and Kraut, J. (1987) Yeast cytochrome c peroxidase: Mutagenesis and expression in Escherichia coli show tryptophan-51 is not the radical site in compound I. Biochemistry, 26, 351-360. doi:10.1021/bi00376a004
- Smith, A.T., Santama, N., Decay, S., Edward, M., Bray, R.C., Thorneley, R.N.F. and Burke, J.F. (1990) Expression of a synthetic gene for horseradish peroxidase-C inn Escherichia coli and folding and activation of the recombinant protein with Ca2+ and haem. Journal of Biological Chemistry, 265, 13335-13343.
- Doyle, W.A. and Smith, A.T. (1996) Expression of lignin peroxidase H8 in Escherichia coli: folding and activation of the recombinant enzyme with Ca2+ and haem. Biochemistry Journal, 315, 15-19.
- Kim, S.J., Lee, J.A., Won, K., Kim, Y.H. and Song, B.K. (2009) Functional expression of Coprinus cinereus peroxidase in Pichia pastoris. Process Biochemistry, 44, 731-735. doi:10.1016/j.procbio.2009.03.004
- Pere-Boada, M., Doyle, W.A., Ruiz-Duenas, F.J., Martinez, A.T. and Smith, A.T. (2002) Expression of Pleurotus eryngii versatile peroxidase in Escherichia coli and optimization of in vitro folding. Enzyme Microbial Technology, 30, 518-524. doi:10.1016/S0141-0229(02)00008-X
- Whitwam, R.E. and Tien, M. (1996) Heterologous expression and reconstitution of fungal Mn peroxidase. Archives of Biochemistry and Biophysics, 333, 439-446. doi:10.1006/abbi.1996.0413
- Whitwam, R.E., Gazarian, I.G. and Tien, M. (1995) Expression of fungal manganese peroxidase in E. coli and refolding to yield active enzyme. Biochemical and Biophysical Research Communications, 216, 1013-1017. doi:10.1006/bbrc.1995.2721
- Conesa, A., Punt, P.J., and Van Den Hondel, C.A.M.J.J. (2002) Fungal peroxidases: Molecular aspects and applications. Journal Biotechnology, 93, 143-158. doi:10.1016/S0168-1656(01)00394-7
- Punt, P.J. Van Biezen, N., Conesa, A., Albers, A., Mangnus, J., and Van Den Hondel, C.A.M.J.J. (2002) Filamentous fungi as cell factories for heterologous protein production. Trends Biotechnology, 20, 200-206. doi:10.1016/S0167-7799(02)01933-9
- Stewart, P., Whitwam, R.E, Kersten, P.J., Cullen, D. and Tien, M. (1996) Efficient expression of a Phanerochaete chrysosporium manganese peroxidase gene in Aspergillus oryzae. Applied Environmental Microbiology, 62, 860- 864.
- Kishi, K., Kusters-van Someren, M., Mayfield, M.B., Sun, J., Loehr, T.M. and Gold, M.H. (1996) Characterization of manganese(II) binding site mutants of manganese peroxidise. Biochemistry, 35, 8986-8994. doi:10.1021/bi960679c
- Kishi, K., Hildebrand, D.P., Kusters-van Someren, M., Gettemy, J., Mauk, A.G. and Gold, M.H. (1997) Site-directed mutationat phenylalanine-190 of manganese peroxidase: Effects on stability, function, and coordination. Biochemistry, 36, 4268-4277. doi:10.1021/bi962627t
- Whitwam, R., Brown, K.R., Musick, M., Natan, M.J. and Tien, M. (1997) Mutagenesis of the Mn2+-Binding site of manganese peroxidase affects oxidation of Mn2+ by both compound I and compound II. Biochemistry, 36, 9766- 9773. doi:10.1021/bi9708794
- Andrawis, A., Pease, E. and Tien, M. (1990) Biotechnology in Pulp and Paper Manufacture. In: Kirk, T.K. and Chang, H.M., Eds., Applications and Fundamental Investigations. Butterworth-Heinemann, Boston, 601.
- Gelpke, M.D.S., Mayfield-Gambill, M., Cereghino, G.P. and Gold, M.H. (1999) Homologous Expression of Recombinant Lignin Peroxidase in Phanerochaete chrysosporium. Applied Environmental Microbiology, 65, 1670- 1674.
- Sambrook, J., Fritsch, E. and Maniatis, T. (1989) Molecular cloning: A Laboratory Manual. Corporation for Supportive Housing, New York.
- Doyle, W.A., Blodig, W., Veitch, N.C., Piontex, K. and Smith, A.T. (1998) Two substrate interaction sites in lignin peroxidase revealed by site-directed mutagenesis. Biochemistry, 37, 15097-15105. doi:10.1021/bi981633h
- Tien, M. and Kirk, K.I. (1988) Lignin peroxidase of Phanerocaete chrysosporium. Methods Enzymology, 161, 238-249. doi:10.1016/0076-6879(88)61025-1
- Wariishi, H., Valli, K. and Gold, M.H. (1992) Manganese peroxidase from the basidiomycete Phanerochaete chrysosporium. Kinetic mechanism and role of chelators. Journal Biology Chemistry, 267, 238-249.
- Gold, M.H., Kuwahara, M., Chiu, A.A. and Glenn, J.K. (1984) Purification and characterization of an extracellular H2O2-requiring diarylpropane oxygenase from the whiterot basidiomycete Phanerocaete chrysosporium. Archives of Biochemistry and Biophysics, 234, 353-362. doi:10.1016/0003-9861(84)90280-7
- Dunford, H.B. (1982) Peroxidases. Advances in Inorganic Biochemistry. Wiley-Vch, New York, 4, 41-68.
- Dunford, H.B. (1991). Horseradish peroxidase: Structure and kinetic properties, Peroxidses in chemistry and biology, CRC Press, Boca Raton.
- Dunford, H.B. (1999) Spectroscopy of horseradish peroxidase. I: Optical, Resonance Raman, Magnetic circular dichroism, X-ray absorption, and Diffraction. Wiley-Vch, New York.
- Wang, Y., Vazquez-Duhalt, R. and Pickard. M.A. (2002). Purification, characterization, and chemical modification of manganese peroxidase from Bjerkandera adusta UAMH 8258. Current Microbiology, 45, 77-87. doi:10.1007/s00284-001-0081-x
- Johjima, T., Itoh, N., Kabuto, M., Tokimura, F., Nakagawa, T., Wariishi, H. and Tanaka, H. (1999) Direct interaction of lignin and lignin peroxidase from Phanerochaete chrysosporium. Proceedings of National Academy of Science, 96, 1989-1994. doi:10.1073/pnas.96.5.1989
- Teske, J.G., Savenkova, M.I., Mauro, J.M., Erman, J.E. and Satterlee, J.D. (2000) Yeast cyrochrome c peroxidase expression in Escherichia coli and rapid isolation of various highly pure holoenzymes. Protein expression and purification, 19, 139-147. doi:10.1006/prep.2000.1220
- Matsubara, M., Suzuki, J., Deguchi, T., Miura, M. and Kitaoka, Y. (1996) Characterization of manganese peroxidases from the hyperlignolytic fungus IZU-154. Applied Environmental Microbioogy, 62, 4066-4072.
- Schneegab, I., Hofrichter, M., Scheibner, K. and Fritsche, W. (1997) Purification of the main manganese peroxidase isozyme MnP2 from the white-rot fungus Nematoloma frowardii b19. Applied Microbiology and Biotechnology, 48, 602-605. doi:10.1007/s002530051102