American Journal of Molecular Biology
Vol.2 No.2(2012), Article ID:18949,13 pages DOI:10.4236/ajmb.2012.22016
Tandem repetitions in transcriptomes of some Solanaceae species
![]()
1University School of Biotechnology, Guru Gobind Singh Indraprastha University, Dwarka, New Delhi, India
2Defence Institute of Bio-Energy Research, Goraparao, Haldwani, India
Email: prof.pcsharma@gmail.com
Received 26 January 2012; revised 13 March 2012; accepted 26 March 2012
Keywords: ESTs; Solanaceae; Tandem Repeats; Transcriptome; TRF
ABSTRACT
Characterization of occurrence, density and motif sequence of tandem repeats in the transcribed regions is helpful in understanding the functional significance of these repeats in the modern genomes. We analyzed tandem repeats present in expressed sequences of thirteen species belonging to genera Capsicum, Nicotiana, Petunia and Solanum of family Solanaceae and the genus Coffea of Rubiaceae to investigate the propagation and evolutionary sustenance of these repeats. Tandem repeat containing sequences constituted 1.58% to 7.46% of sequences analyzed. Tandem repetitions of size 2, 15, 18 and 21 bp motifs were more frequent. Repeats with unit sizes 21 and 22 bp were also abundant in genomic sequences of potato and tomato. While mutations occurring in these repeats may alter the repeat number, genomes adjust to these changes by keeping the translated products unaffected. Surprisingly, in majority of the species under study, tandem repeat motif length did not exceed 228 bp. Conserved tandem repeat motifs of sizes 180, 192 and 204 bp were also abundant in the genomic sequences. Our observations lead us to propose that these tandem repeats are actually remnants of ancestral megasatellite repeats, which have split into multiple repeats due to frequent insertions over the course of evolution.
1. INTRODUCTION
The extent of repetitiveness in nucleotide base sequences varies remarkably across genomes and generally exceeds the statistically derived expected values [1]. Taking into account some direct and indirect influence on the survival of the organism [2,3], it is not unusual to expect repetitive DNA constituting major portion of the present day genomes [4,5]. Tandem repeats are ubiquitous in a broader sense as they occur at telomeres, centromeres, genic regions, intergenic regions and even at interspersed sites [6]. A deeper analysis of the eukaryotic genomes suggests a non-random distribution of tandem repeats [5, 7]. Comparative genomics focusing on the tandem repeats lying within or close to genes helps in understanding the functional significance of these repeats in modern genomes. Comprehensive experiments involving tandem repeats may be instrumental in generating valuable information about various other biological features related to C-value paradox, organization and evolution of genomes, transcription, etc. [5].
Genome analysis of a number of plant species representing the important family Solanaceae has revealed striking similarities in terms of gene content and organization [8,9]. The wealth of sequence information pertaining to the members of Solanaceae has expanded rapidly in recent times. Currently, genome projects are underway for many members of the Solanaceae including Capsicum annuum (Pepper), Nicotiana benthamiana (Benthamiana tobacco), Nicotiana tabacum (Tobacco), Solanum bulbocastanum, Solanum demissum (Hexaploid Mexican wild potato), Solanum lycopersicoides (Wild nightshade), Solanum lycopersicum (Tomato), Solanum melongena (Brinjal), Solanum peruvium (Wild tomato) and Solanum tuberosum (Potato) (see database “genome projects” at http://www.ncbi.nlm.nih.gov/genomeprj/?term=Solanaceae). Such sequence resources provide an opportunity to get insights into the evolutionary history of closely related species. That is, if the sequences are identical between two species, chances are that the two species might have diverged from each other fairly recently. Points of disagreement in the sequence homology indicate a longer evolutionary distance between the given species, also reflected in their taxonomic positions. These lines are explored in this paper by comparative analysis of the organization and distribution of tandem repeats in unigenes and EST sequences of thirteen members of family Solanaceae and two members of a closely related family, Rubiaceae. We believe that such studies will be helpful in addressing some of the most interesting questions in the field of genomics and transcriptomics concerning the patterns and significance of tandem repetition of sequences, and the factors that maintain and propagate these tandem repeats over the generations.
2. METHODS
2.1. Sequence Resources and Initial Processing
The unigene sequences of potato, tomato and tobacco were downloaded from unigene database of NCBI. Similarly, EST sequence data for twelve species (Table 1) were downloaded from dbEST of NCBI (http://www.ncbi.nlm.nih.gov/nucest/). All the sequence data were downloaded in fasta format. ESTs were clustered using the CAP3 program [10]. Subsets of this data were further randomly clustered based on sequence homology using the standalone version of BLASTn at various stages during the study. The purpose of including the latter step was to construct cross-species clusters of EST-SSRs. NCBI descriptions thus obtained were retained for the best hit as long as E-value was less than 1e–10 and alignment score was >200.
In addition, 5 Mb and 90 Mb of potato and tomato genomic sequences, respectively, available in the public domain were also analyzed for the presence of tandem repeats.
2.2. Identification of Tandem Repeats and Cross-Species Comparisons
The identification of tandem repeats was performed by using the search tool Tandem Repeats Finder [11] according to the parameter value scores of 2, 7, 7, 80, 10, 50 and 500 for match, mismatch, indels, matching probability, indel probability, minimum alignment score and maximum period size, respectively. As TRF detects more than one repeat on the basis of alignment score at the same site, we rectified this anomaly by only recognizing the repeat with smallest motif. Wherever there was a tie on the basis of motif size, longer sequence was considered. If the tie was observed in terms of length span also, then lower entropy was given a preference. As entropy stands for randomness in thermodynamics, higher entropy would mean randomness (or less orderliness in the sequence of nucleotides) in terms of sequence analysis. Lower entropy automatically means ordered occurrence of nucleotides, thereby leading to the formation of repeats. Repeats with motif size of 2 - 6 bp were identified as microsatellites and rest of the sequences were termed as minisatellites. Considering the fact that a number of stretches of (A/T)n would actually be non genomic poly-A tails, mononucleotide repeats were excluded from the present analysis, if they occurred in the end of the sequences. The microsatellite repeats were grouped into

Table 1. Summary of dataset analyzed for the occurrence of tandem repeats in transcriptomic sequences of Solanaceae and the extent of repetitiveness present.
different classes according to Jurka and Pethiyagoda [12].
To predict the cross-species transferability of these repeats, all the sequences were also scanned by VNTRfinder [13]. This exercise limited the output only to the PCR amplifiable transferable repeats showing length polymorphism, when compared with another species. The conservation of repeats across the species was also studied using BLASTn according to the parameters described above.
Synteny mapping between potato and tomato contigs was carried out using glocal algorithm [14] in Vista Genome Browser (http://pipeline.lbl.gov/cgi-bin/gateway2) [15] in an all versus all patterns. Output of genome vista browser was retrieved through e-mail.
3. RESULTS
3.1. Abundance of Tandem Repeats in Solanaceae Transcriptomes
Occurrence of tandem repeats in the transcriptomes analyzed showed variation on different accounts depending upon the species concerned. As evident from Table 1, tandem repeat containing sequences ranged from a minimum of 1.58% in Nicotiana sylvestris to a maximum of 7.46% sequences in Petunia axillaris. In terms of transcriptome coverage, most of the species showed 0.5 - 0.6% of sequences harbouring tandem repeats (Table 1). The average GC content of tandem repeated sequences remained ~ 41%. Among the tandem repeats with longer motifs, mononucleotide A was the most common followed by T.
Minisatellite repeats essentially occurred either in the exonic regions or overlapped with the exonic regions. Tandem repeats with smaller unit size, in general, were more abundant than the repeats with longer repeat unit. Interestingly, a marked dominance of tandem repeats with repeat unit size (bp) in the multiple of three was noticed (Figure 1). In fact, 64% of all the repeats identified in this study showed this characteristic. Among all the repeats mined, repeats with motif size of 2, 15, 18 and 21 bp were more abundant. Tandem repeats with repeat unit size of 2 bp were extraordinarily abundant in Capsicum constituting 14% of all the tandem repeats. Interestingly, 27% of all the dinucleotide repeats reported in Solanaceae transcriptomes under study originated from Capsicum sequences. A similar dominance of dinucleotide repeats was also prevalent in Coffea canephora. Repeats of unit sizes 21 and 22 bp also represented the most abundant tandem repeats in genomic sequences of potato and tomato, and also in rice and humans (our unpublished data). When the repeat richness of unigene sequences was compared with genomic sequences in potato and tomato, no definite trend could be observed, except that a higher frequency of tandem repeats was observed in genomic sequences. The repeats with unit size ranging from 15 to 22 bp were markedly more abundant in genomic sequences as seen in Figure 2. Evidently, tandem repeats with motif sizes between 7 and 30 bp account for the maximum number of loci and longer arrays both in the genomic as well as transcribed sequences of Solanaceae.
3.2. Cross-Species Comparisons
While the cross-species conservation within a genus was more visible (Table 2), the probability of finding an orthologue in a different genus was quite low. For many tandem repeats, the encoded repetitive peptide sequence was found longer than that expected using ORFpredictor
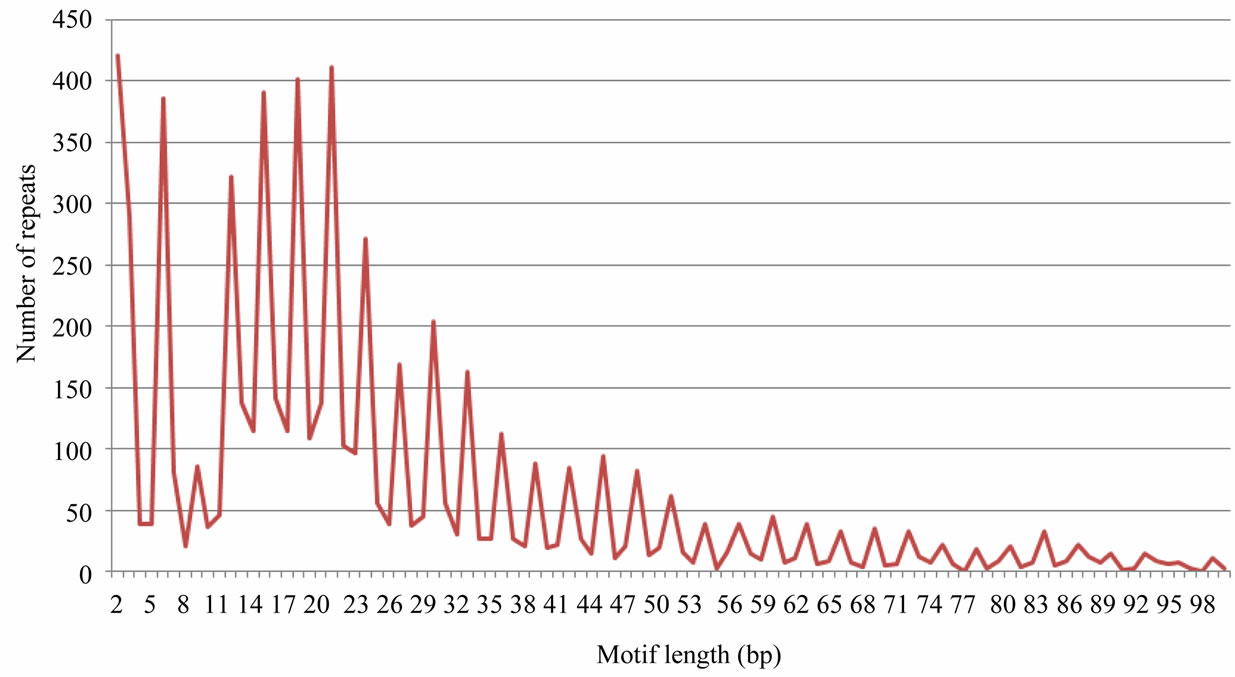
Figure 1. Abundance of tandem repeats with different repeat units in Solanaceae.
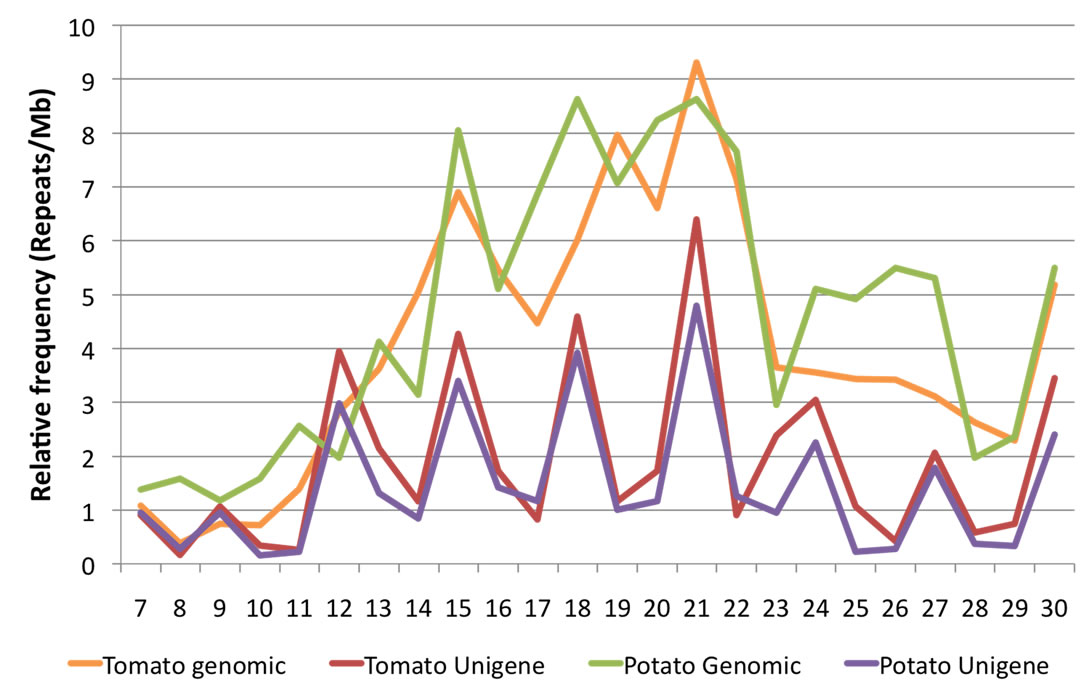
Figure 2. Occurrence of tandem repeats in unigene and genomic sequences of tomato and potato. A different pattern of distribution of genomic versus transcriptomic tandem repeats with motif sizes 7 bp - 30 bp is clearly visible.
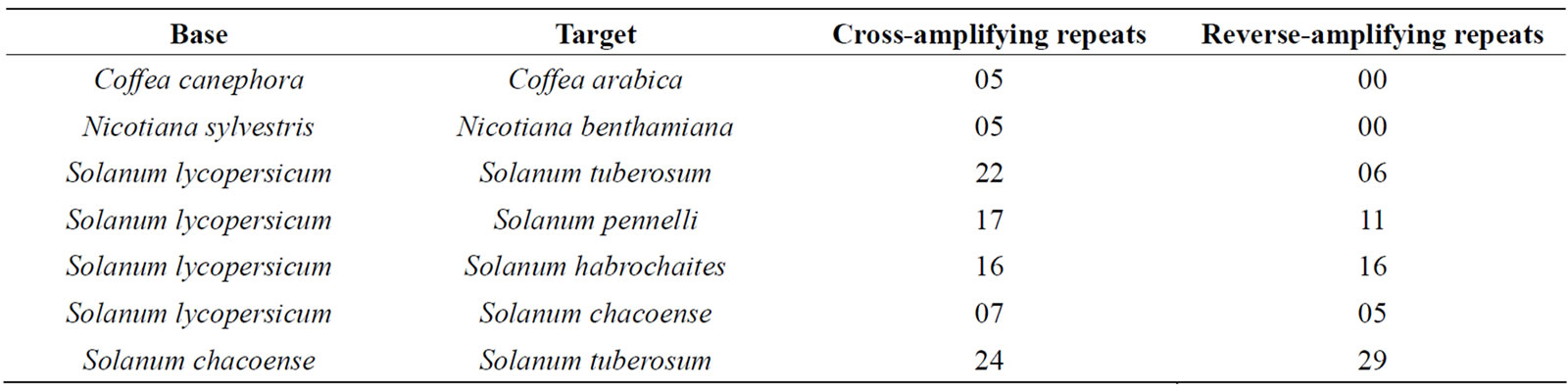
Table 2. Cross-species PCR transferability in Solanaceae, as predicted by VNTRfinder.
(data not shown). Interestingly, more number of orthologous pairs of tandem repeats were observed using BLASTn than predicted by ePCR module of VNTRfinder. For example, in the tomato-potato pair, more than 50% of the microsatellite containing sequences had an orthologous match in the other species database, however, not all of those contained a microsatellite. A similar observation was drawn for the N. tabacum and N. benthamiana pair. As the VNTRfinder predicts the cross-species PCR amplification based on a number of parameters and not merely the sequence similarity, it is quite possible that most of the orthologues fail to cross-amplify under optimal PCR conditions. Although the exact composition of a tandem repeat could not be traced in the orthologous sequences in some instances, but considerable sequence similarity and the reading frame may still be preserved. With the available data, it was not possible to conclude which of the alleles among the orthologues was the ancestral one. Identifcation and study of a common ancestor (or its direct descendent) could be partially useful. Synteny mapping between tomato and potato genomes for tandem repeats revealed different trends, for examplefew of the tandem repeats were conserved between the two genomes, while others were found showing variations in the otherwise conserved genomic regions. The mapped synteny was not absolute and indels as well as micro-inversions have frequently occurred since the divergence of potato and tomato (Figure 3). The overall repetitive sequence content in potato and tomato was comparable in terms of the genomic coverage (Figure 2). Most of these tandem repeats could not be characterized, as except for a single instance of accumulation of telomeric/centromeric heptanucleotide repeats, no other telomeric or centromeric repeats could be identified.
3.3. Tandem Repeat Richness and Motif Length
While searching for tandem repeats in this study, we had set an upper length limit of 500 bp for motif size. Surprisingly, in majority of the species, tandem repeat motif length did not exceed 228 bp. Further, among all the repeats with unit length longer than 100 bp, repeats with unit lengths in the multiple of 114 (114×) and particularly 228 bp were most abundant. As shown in Figure 4,
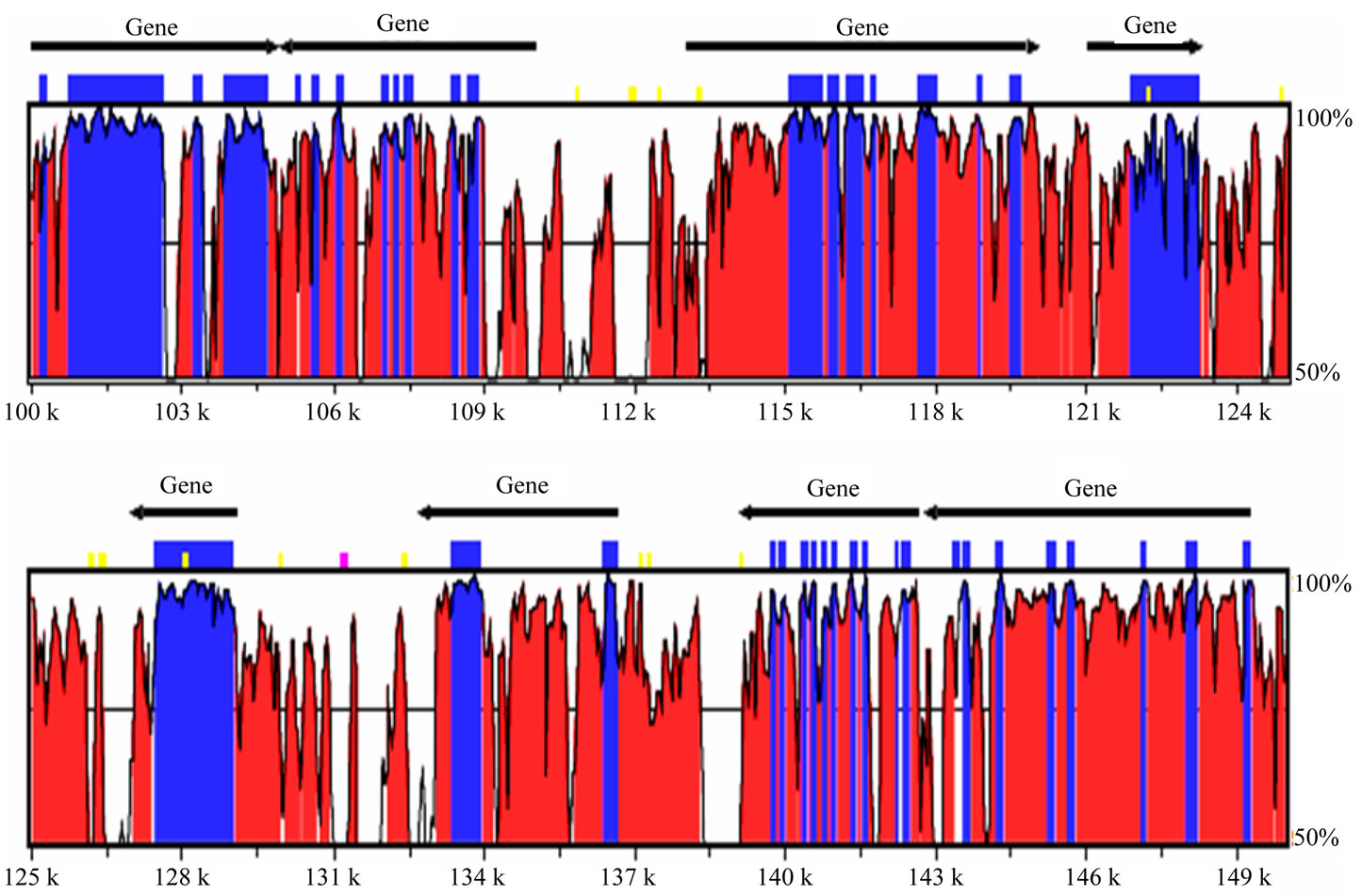
Figure 3. A small region displaying degree of synteny between tomato and potato genomes and various repetitive sequences present in this region.

Figure 4. Repeat unit sizes of longest repeat in different species. Noticeably, 228 bp is a preferred length among the longer repeats in majority of the species.
except for Coffea arabica and Petunia × hybrida, the longest repeat belonged to 114× category. Moreover, repeats with unit length 228 bp, not only showed a marked abundance among the repeats with longer motifs, but also spanned much longer in length (Figure 5).
Interestingly, repeats belonging to the family 114× could not be traced into genomic sequences of potato and tomato, indicating that they were split over two or more exons. Repeats with motif sizes 180, 192 and 204 bp were more abundant in genomic sequences. Similar abundance of 180 bp and 192 bp motif size long tandem repeats was also seen in rice (our unpublished results), and by using BLAST, such repeats were annotated as transposable element proteins. Another interesting feature
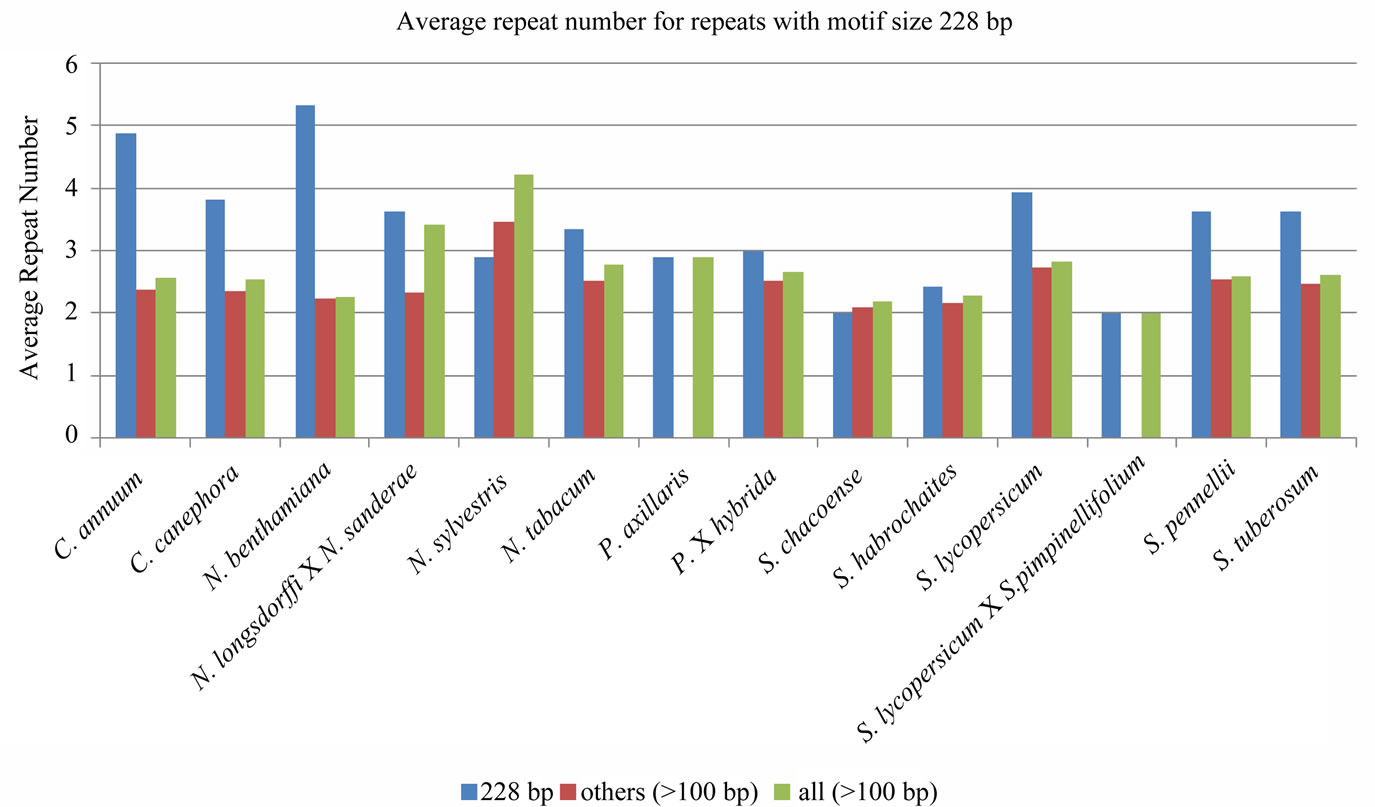
Figure 5. Average repeat number for all the tandem repeats with repeat unit length higher than 100 bp in different species.
of genomic contigs of potato and tomato revealed a marked accumulation of tandem repeats with same sized motif lengths causing a significant deviation in the values of mean and mode of repeat lengths within these contigs (Table 3). Following sequence comparison of the repeat units of these tandem repeats, a high level of similarity (>90% identities in the aligned sequences) was observed.
4. DISCUSSION
Tandem repeats represent a considerable proportion, and yet remain a poorly understood component of the eukaryotic genomes. Opinions differ on their structural and functional significance in the genomes [3]. Various roles have been proposed for tandem repeats highlighting their effect on chromatin organization, crossing over, regulation of gene activity, etc. [16]. Some data is available on the distribution of microsatellites in various genomes [6, 17], but virtually no information is available till date on genomic distribution of minisatellites and satellites. Our experience of working with microsatellites, i.e., tandem repeats with shorter repeat motifs [7,18] suggests that the structure of tandem repeats may be regulated by their neighbouring components of the genome, as also reported for their mutability [19]. However, coding and non coding regions of a genome are regulated by differrent constraints and thus the fine genomic environment at these sites differs from one another. On the same lines, repeated sequence motifs are tolerated in transcriptomes obviously in accordance with the requirements of the ultimate products in the system. Study of tandem repeats present in the transcribed sequences thus makes an interesting area of contemporary research. In the present study, following dynamics and conservation of tandem repeats in genic regions of some members of Solanaceae, we obtained certain interesting insights about their existence in transcriptomic sequences, previously not reported on this scale and also on periodicities in the anticipated protein sequences. The frequency with which tandem repeats occur in ESTs offers a new area for exploration due to the associated translation into protein sequences and thereby providing different abilities to the proteome of an organism.
In the present study, we found that the repeat containing transcriptomic sequences are slightly lesser than what have been reported earlier, and also slightly lesser than the genomic coverage values for angiosperms [5-7]. Poor GC content of tandem repeats might be reflected in the functional utility of these tandem repeats. For example, repeat motifs AG and AAG generally occur in the 5’- UTR regions of the genes and have been suggested to form non-B-DNA, potentially playing important roles in the regulation of gene activity [20]. (CTT)n repeats, complementary to (AAG)n, are also potential sites of cytosine methylation, and therefore, provide candidate sites for inhibiting transcription elongation in plants [21,22]. Hypervariability of these regions in exonic regions might lead to novel amino acid sequences that may in some cases lead to several disorders, as known widely in humans [23]. Nevertheless, till date no specific function could be assigned to amino acid sequence expansions





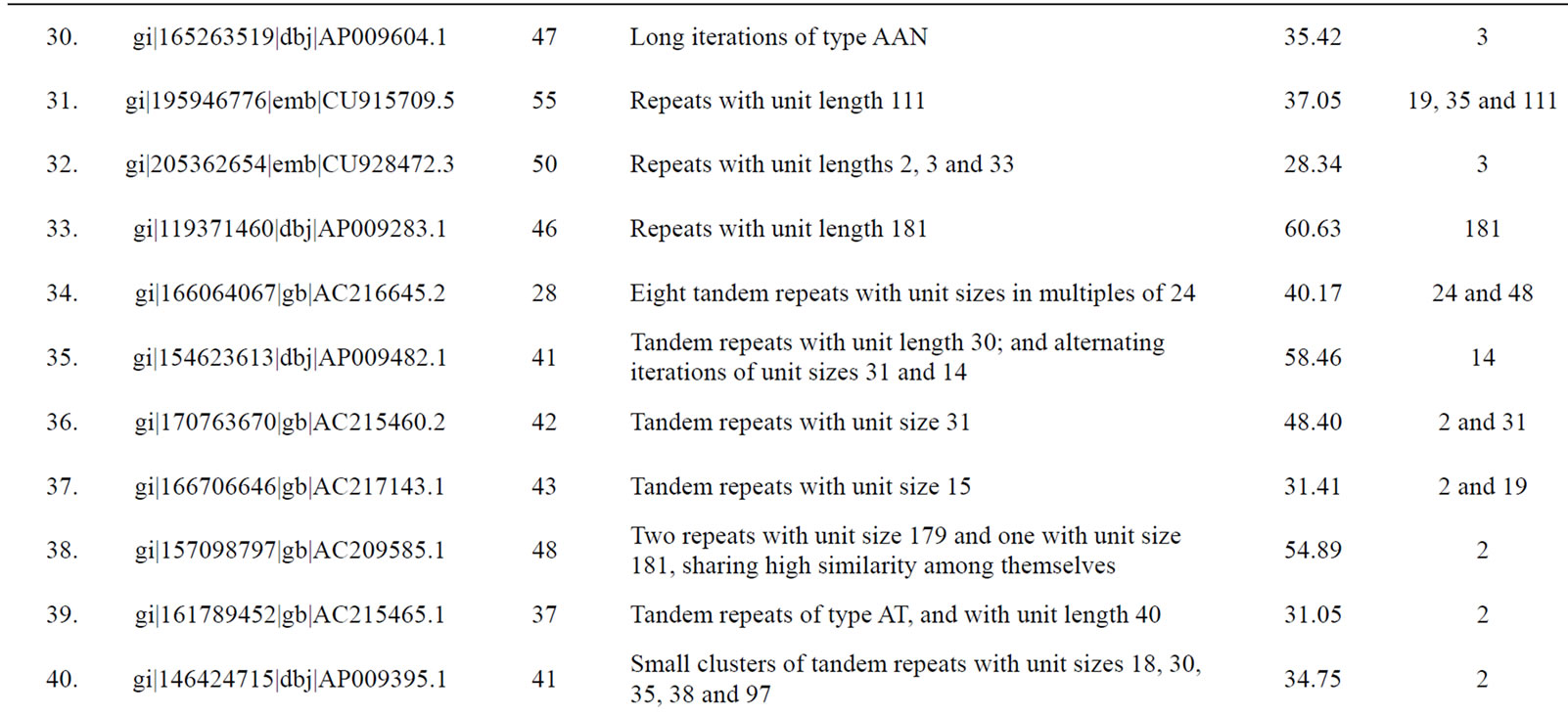


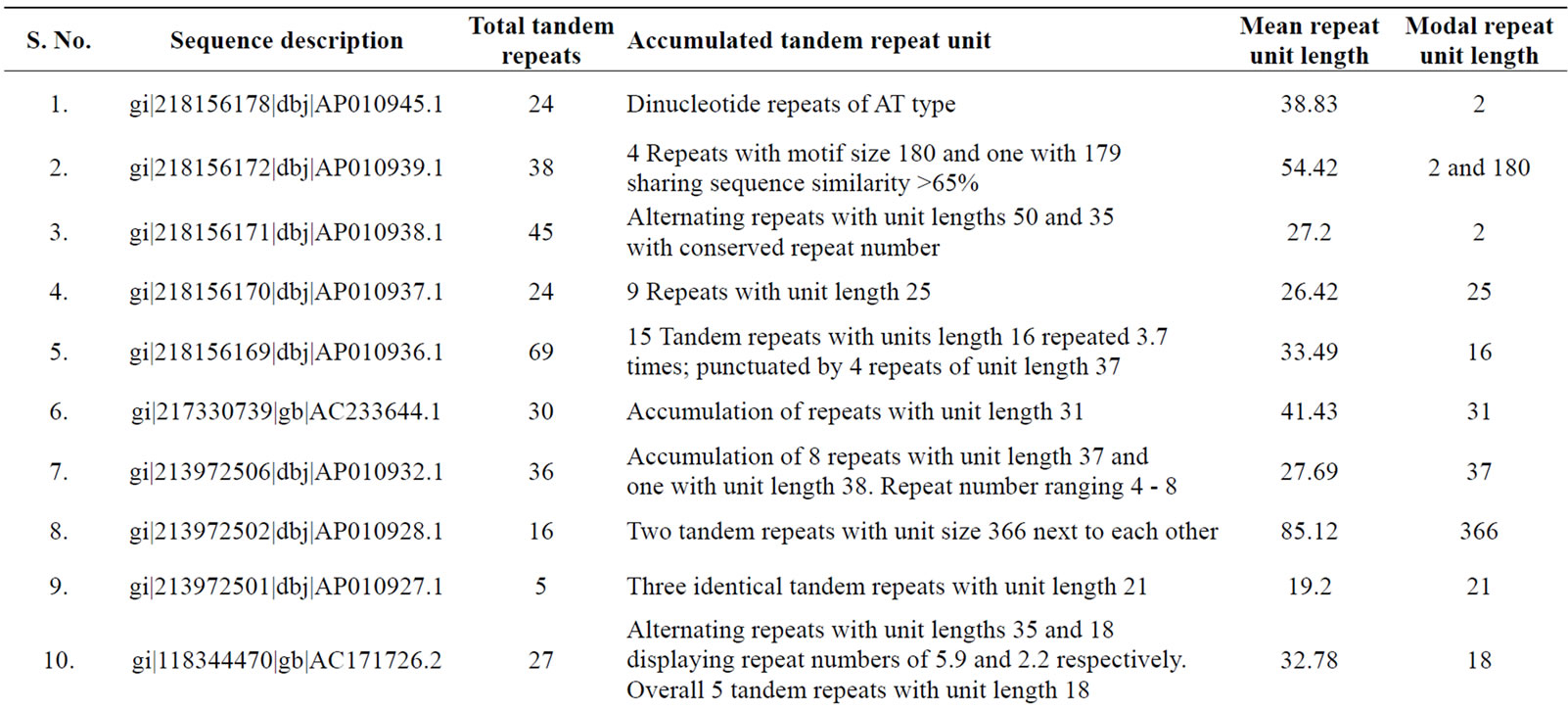
Table 3. Tomato genomic contigs showing accumulation of tandem repeats with similar repeat units.
and it would not be unwise to think that this process might be a contributor to the evolution of newer genes. De Grassi and Ciccarelli [24] on the basis of their studies on “internal tandem repeats” in genes lying in duplicated regions of human genome observed that modifications in tandem repeats always occurred in terminal exon of the genes. The event is favourable, as this would not affect the original composition of proteins [24], and will make the gene available for alternative splicing. In fact, the effect of polymorphisms at tandem repeat sites on gene expression is slowly getting established [23], even if the tandem repeat polymorphism is generally confined to introns [25]. When the tandem repeats occur at intron-exon boundry, novel introns may be formed due to modifications of their length or sequence, leading to formation of alternative transcripts [24,26].
The marked dominance of tandem repeats with unit lengths in multiples of three may be considered as an extension of the observation that trinucleotide repeats are predominantly present in genic sequences, particularly in exons [7,27]. However, such an observation also contrasts the trend seen in the genomic sequences where the abundance linearly falls with increasing length of the repeat unit size [5]. Predominance of repeats with unit size 2, 15, 18 and 21 bp was interesting. While occurrence of dinucleotide repeats in 5’-UTR could be explained by their expected participation in the transcription machinery as transcription factor binding sites [20], more intriguing was the abundance of tandem repeats with repeat unit size of 21 bp as the second most abundant class in transcriptomes under study (Figure 1). However, given the universality of such abundance, we believe that they have some important function, for which they are retained in the genomes. In general, tandem repeats with unit lengths in multiples of three are more abundant in genomes and certain genomic forces have facilitated their longer iterations. Nevertheless, abundance of tandem repeats with unit sizes 7 - 30 bp over the longer ones is in accordance with that reported earlier by Navajas-Perez and Patterson [5] for other plant genomes. Brandstorm et al. [28] suggested that these sequences serve as hot spots of recombination. Sharma and Raina [29] also demonstrated that tandem repeats of various types represent species-specific and chromosomespecific heterochromatin patterns.
Conservation of tandem repeats and their evolution in plant genomes is likely to be dictated by the features such as the length and sequence of the basic repeat unit [30]. However, Richard and Dujon [31] also reported the transferability of minisatellites across genera. Thus, despite prevalent insertions, deletions and substitution events, tandem repeats in genes are still under positive natural selection. Evidences in support of such proposition are made available from studies in humans [32,33]. Jordan et al. [34] also endorsed similar observations and conclusions on the basis of their cross-species comparisons in Neisseria spp., and suggested the significance of this phenomenon in providing adaptability to the host. This view later also got support from Verstrepen et al. [35] and Levdansky et al. [36] following their studies in yeast and Aspergillus fumigatus, respectively.
A combination of polymerase slippage and point mutations [37] can either elongate or shorten a tandem repeat. A longer allele, if considered ancestral, can get shortened in two wayseither a mutation event occurs at one of the ends of the locus thereby reducing the repeat number of the locus or a mutation occurring in the middle of a locus breaking the locus into two smaller loci. If a shorter allele is considered ancestral, it can get elongated either by the joining of two nearby loci or by increasing its length by one repeat at one time [38]. Tandem repeats have probably undergone a complicated set of mutational events altering their length and have maintained high mutation rates even in expressed regions [39]. Trifonov [40] suggested that microsatellites in genes have an adaptive advantage against stress conditions. Longer repeat sequences modulate the expression of genes under stress. ![]() The ESTs harboring microsatellites, and those where a cross-generic orthologue is conserved, might have a range of functions such as coding for signaling proteins, kinases or transcription factors or a MADS box gene. Fujimori et al. [41] found 46.5% of translation-related housekeeping genes in plants having a microsatellite region in their predicted 5’-UTR. Microsatellite repeats in untranslated regions probably regulate gene expression by making certain DNA-protein interactions [42,43]. While the mutations occurring in these repeats may reduce the repeat number, genomes adjust to these changes by keeping the translated products unaffected. Since their occurrence is prevalent in conserved housekeeping genes, it is suggested that these repeats might have been inherited from a common ancestor and due to vitality of their functions; these repeats or their remnants can distinctly be identified. We do not over rule the possibility of harbouring mutations in these genes by organisms in response to ecological or environmental stress, as each of these species has faced different environmental and domestication requirements. These issues probably require further investigations in vertical lineage instead of horizontal comparisons among different species.
The ESTs harboring microsatellites, and those where a cross-generic orthologue is conserved, might have a range of functions such as coding for signaling proteins, kinases or transcription factors or a MADS box gene. Fujimori et al. [41] found 46.5% of translation-related housekeeping genes in plants having a microsatellite region in their predicted 5’-UTR. Microsatellite repeats in untranslated regions probably regulate gene expression by making certain DNA-protein interactions [42,43]. While the mutations occurring in these repeats may reduce the repeat number, genomes adjust to these changes by keeping the translated products unaffected. Since their occurrence is prevalent in conserved housekeeping genes, it is suggested that these repeats might have been inherited from a common ancestor and due to vitality of their functions; these repeats or their remnants can distinctly be identified. We do not over rule the possibility of harbouring mutations in these genes by organisms in response to ecological or environmental stress, as each of these species has faced different environmental and domestication requirements. These issues probably require further investigations in vertical lineage instead of horizontal comparisons among different species.
Occurrence and abundance of repeats with longer units also raised curiosity. According to De Grassi and Cicarrelli [24] tandem repeats with 30 bp repeat units prevailing at least four times more frequently causing modifications in human genes in duplicated regions of the genome. Tandem repeats with longer units according to De Grassi and Ciccarelli [24] are more variable than repeats
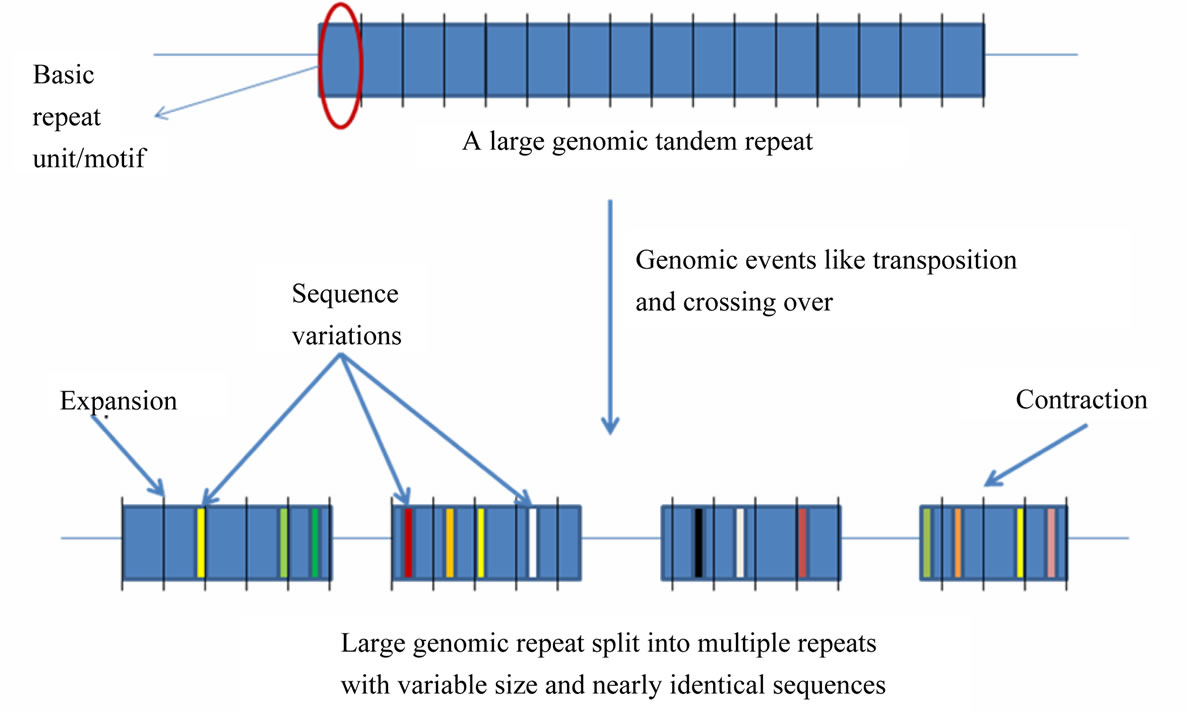
Figure 6. A generalized mechanism leading to “accumulated” tandem repeats with identical repeat unit from an ancestral “mega-satellite” of the past. This leads to the accumulation of repeats with similar size in genomic contigs.
with higher repeat number. If translated, these repeats would induce periodicities in protein structures. This might well be a possible situation exploited by the cellular machinery in preferring single subunit proteins that play the roles of multi-subunit proteins. The energetics and kinetics of TR-containing proteins provide new insights into folding rates and protein stability [44]. The understandable benefit of a single subunit protein is its ensured availability independent of stoichiometry. In fact, presence of tandem repeats in protein sequences is well recorded [39] with most of them displaying a smaller repeat unit of 5 - 20 amino acids. Repeated domains in proteins are known to be associated with a variety of functions [39]. Kashi and King [3] also suggested that repeated sequences may result in open reading frames (ORFs) of substantial length, integrated into an actively transcribed region. Richard and Dujon [31] reported minisatellites containing genes to be associated with genes encoding cell wall proteins.
Since a high level of similarity was observed in the sequence of clustered repeats with long units (those which were discovered from the same contigs), we propose that these tandem repeats are actually remains of an ancestral megasatellite repeat, which has split into multiple repeats due to frequent insertions during the course of evolution. Each of the broken unit too has accumulated a number of indels and substitutions over a period of time downgrading them to “nearly identical” to each other from “identical” units of the past. A generalized mechanism creating such accumulation of repeats in genomic regions is depicted in Figure 6.
There have been certain suggestions that tandem repeats might have served as a mode for evolution of novel genes [24,45], simply by altering the number of times a sequence motif is repeated. In the process, tandem repeats might have contributed to the fitness of the organism in the prevailing environment. Marcotte et al. [46] suggested that repeat expansion shaped many protein domain families like leucine rich repeats family, and this is an important mode of evolution of eukaryotic genomes [47]. Vergnaud and Denoeud [48] used the method similar to ours, but different definition to analyze minisatellites in human chromosome 22, Arabidopsis thaliana chromosome 4, and Caenorhabditis elegans chromosome 1 by the use of the TRF software and reported the preferential occurrence of these repeats near telomeric and centromeric regions of the genomes. Richard et al. [43], however, maintained that there is no such bias, when complete genomic sequences are analyzed. Nevertheless, at this stage any of the conclusions would be pre-mature as minisatellites are less studied genomic constituents than microsatellites [49,50].
5. ACKNOWLEDGEMENTS
Authors are thankful to Indian Council of Agricultural Research (ICAR), Defence Research and Development Organization (DRDO) and Department of Science and Technology (DST), Government of India for providing research grants to support their research on microsatellites. Ankit Jain assisted in the formating of the manuscript.
REFERENCES
- Haubold, B. and Wiehe, T. (2006) How repetitive are genomes? BMC Bioinformatics, 7, 541. doi:10.1186/1471-2105-7-541
- Shaprio, J.A. and von Stanberg, R. (2005) Why repetitive DNA is essential to genome function? Biological Reviews of the Cambridge Philosophical Society, 80, 227-250. doi:10.1017/S1464793104006657
- Kashi, Y. and King, D.G. (2006) Simple sequence repeats as advantageous mutators in evolution. Trends in Genetics, 22, 253-259. doi:10.1016/j.tig.2006.03.005
- Piegu, B., Guyot, R., Picault, N., et al. (2006) Doubling genome size without polyploidization: Dynamics of retrotransposition-driven genomic expansions in Oryza australiensis, a wild relative of rice. Genome Research, 16, 1262-1269. doi:10.1101/gr.5290206
- Navajas-Perez, R. and Patterson, A. (2009) Patterns of tandem repetition in plant whole genome assemblies. Molecular Genetics and Genomics, 281, 579-590. doi:10.1007/s00438-009-0433-y
- Sharma, P.C., Grover, A. and Kahl, G. (2007) Mining microsatellite repeats in eukaryotic genomes. Trends in Biotechnology, 25, 490-498. doi:10.1016/j.tibtech.2007.07.013
- Grover, A., Aishwarya, V. and Sharma, P.C. (2007) Biased distribution of microsatellite motifs in the rice genome. Molecular Genetics and Genomics, 277, 469-480. doi:10.1007/s00438-006-0204-y
- Mueller, L.A., Solow, T.H., Taylor, N., et al. (2005) The SOL Genomics Network: A comparative resource for Solanaceae biology and beyond. Plant Physiology, 138, 1310-1317. doi:10.1104/pp.105.060707
- Bombarely, A., Menda, N., Tecle, I.Y., et al. (2011) The sol genomics network (solgenomics.net): Growing tomatoes using Perl. Nucleic Acids Research, 39, D1149- D1155. doi:10.1093/nar/gkq866
- Huang, X. and Madan, A. (1999) CAP3: A DNA sequence assembly program. Genome Research, 9, 868-877. doi:10.1101/gr.9.9.868
- Benson, G. (1999) Tandem repeats finder: A program to analyze DNA sequences. Nucleic Acids Research, 27, 573-580. doi:10.1093/nar/27.2.573
- Jurka, J. and Pethiyagoda, C. (1995) Simple repetitive DNA sequences from primates: Compilation and analysis. Journal of Molecular Evolution, 40, 120-126. doi:10.1007/BF00167107
- O’Dushlaine, C.T. and Shields, D.C. (2006) Tools for the identification of the variable and potentially variable tandem repeats. BMC Genomics, 7, 290. doi:10.1186/1471-2164-7-290
- Brudno, M., Malde, S., Poliakov, A., et al. (2003) Glocal alignment: Finding rearrangements during alignment. Bioinformatics, 1, i54-i62. doi:10.1093/bioinformatics/btg1005
- Frazer, K.A., Pachter, L., Poliakov, A., et al. (2004) VISTA: Computational tools for comparative genomics. Nucleic Acids Research, 32, W273-W279. doi:10.1093/nar/gkh458
- Li, Y.C., Korol, A.B., Fahima, T. and Nevo, E. (2004) Microsatellites within genes: Structure, function and evolution. Molecular Biology and Evolution, 21, 991-1007. doi:10.1093/molbev/msh073
- Zhang, L., Yuan, D., Yu, S., et al. (2004) Preference of simple sequence repeats in coding and non-coding regions of Arabidopsis thaliana. Bioinformatics, 20, 1081- 1086. doi:10.1093/bioinformatics/bth043
- Roorkiwal, M., Grover, A. and Sharma, P.C. (2009) Genome-wide analysis of conservation and divergence of microsatellites in rice. Molecular Genetics and Genomics, 282, 205-215. doi:10.1007/s00438-009-0457-3
- Brock, G.J.R., Anderson, N.H. and Monckton, D.G. (1999) Cis-acting modifiers of expanded CAG/CTG triplet repeat expandability: Associations with flanking GC content and proximity to CpG islands. Human Molecular Genetics, 8, 1061-1067. doi:10.1093/hmg/8.6.1061
- Ng, T.K., Lam, C.Y., Lam, D.S.C., et al. (2009) AC and AG dinucleotide repeats in the PAX6 P1 promoter are associated with high myopia. Molecular Vision, 15, 2239- 2248.
- Hohn, T., Corsten, S., Ricke, S. and Rothnie, H. (1996) Methylation of coding region alone inhibits gene expression in plant protoplasts. Proceedings of the National Academy of Sciences of the United States of America, 93, 8334-8339. doi:10.1073/pnas.93.16.8334
- Colot, V. and Rossignol, J.L. (1999) Eukaryotic DNA methylation as an evolutionary device. BioEssays, 21, 402-411. doi:10.1002/(SICI)1521-1878(199905)21:5<402::AID-BIES7>3.0.CO;2-B
- Jeong, Y.H., Kim, M.C., Ahn, E.-K., et al. (2007) Rare exonic microsatellite alleles in MUC2 influence susceptibility to gastric carcinoma. PloS One, 11, e1163. doi:10.1371/journal.pone.0001163
- De Grassi, A. and Ciccarelli, F.D. (2009) Tandem repeats modify the structure of human genes hosted in segmental duplications. Genome Biology, 10, R137. doi:10.1186/gb-2009-10-12-r137
- Leem, S.H., Londoño-Vallejo, J.A., Kim, J.H., et al. (2002) The human telomerase gene: Complete genomic sequence and analysis of tandem repeat polymorphisms in intronic regions. Oncogene, 21, 769-777. doi:10.1038/sj.onc.1205122
- Catania, F. and Lynch, M. (2008) Where do introns come from? PLoS Biology, 6, e283. doi:10.1371/journal.pbio.0060283
- Varshney, R.K., Graner, A. and Sorrells, M.E. (2005) Genic microsatellite markers in plants: Features and applications. Trends in Biotechnology, 23, 48-54. doi:10.1016/j.tibtech.2004.11.005
- Brandstorm, M., Bagshaw, A.T., Gemmell, N.J. and Ellegren, H. (2008) The relationship between microsatellite polymorphism and recombination hot spots in the human genome. Molecular Biology and Evolution, 25, 2579- 2587. doi:10.1093/molbev/msn201
- Sharma, S. and Raina, S.N. (2005) Organization and evolution of highly repeated satellite DNA sequences in plant chromosomes. Cytogenetic and Genome Research, 109, 15-26. doi:10.1159/000082377
- Ugarkovic, D. and Plohl, M. (2002) Variation in satellite DNA profiles—Causes and effects. EMBO Journal, 21, 5955-5959.
- Richard, G.-F. and Dujon, B. (2006) Molecular evolution of minisatellites in hemiascomycetous yeasts. Molecular Biology and Evolution, 23, 189-202. doi:10.1093/molbev/msj022
- O’Dushlaine, C.T., Edwards, R.J., Park, S.D. and Shields, D.C. (2005) Tandem repeat copy number variation in protein-coding regions of human genes. Genome Biology, 6, R69. doi:10.1186/gb-2005-6-8-r69
- Yu, F., Sabeti, P.C., Hardenbol, P., et al. (2005) Positive selection of a pre-expansion CAG repeat of the human SCA2 gene. PLoS Genetics, 1, e41. doi:10.1371/journal.pgen.0010041
- Jordon, P., Snyder, L.A. and Saunders, N.J. (2003) Diversity in coding tandem repeats in related Neisseria spp. BMC Microbiology, 3, 23. doi:10.1186/1471-2180-3-23
- Verstrepen, K.J., Jansen, A., Lewitter, F. and Fink, G.R. (2005) Intragenic tandem repeats generate functional variability. Nature Genetics, 37, 986-990. doi:10.1038/ng1618
- Levdansky, E., Romano, J., Shadkchan, Y., et al. (2007) Coding tandem repeats generate diversity in Aspergillus fumigatus genes. Eukaryotic Cell, 6, 1380-1391. doi:10.1128/EC.00229-06
- Calabrese, P.P., Durrett, R.T. and Aquadro, C.A. (2001) Dynamics of microsatellite divergence under stepwise mutation and proportional slippage/ point mutation model. Genetics, 159, 839-852.
- Ohta, T. and Kimura, M. (1973) A model of mutation appropriate to estimate the number of electrophoretically detectable alleles in a finite population. Genetics Research, 22, 201-204. doi:10.1017/S0016672300012994
- Naamati, G., Fromer, M. and Linial, M. (2009) Expansion of tandem repeats in sea anemone Nematostella vectensis proteome: A source for gene novelty? BMC Genomics, 10, 593. doi:10.1186/1471-2164-10-593
- Trifonov, E.N. (2003) Tuning function of tandemly repeating sequences: a molecular device for fast adaptation. In: Wasser, S.P., Ed., Evolutionary Theory and Processes: Modern Horizons, Papers in Honour of Eviatar Nevo. Kluwer Academic Publishers, Amsterdam, 1-24.
- Fujimori, S., Washio, T., Higo, K., et al. (2003) A novel feature of microsatellites in plants: A distribution gradient along the direction of transcription. FEBS Letters, 554, 17-22. doi:10.1016/S0014-5793(03)01041-X
- Kashi, Y., King, D. and Soller, M. (1997) Simple sequence repeats as a source of quantitative genetic variation. Trends in Genetics, 13, 74-78. doi:10.1016/S0168-9525(97)01008-1
- Richard, G.F., Kerrest, A. and Dujon, B. (2008) Comparative genomics and molecular dynamics of DNA repeats in eukaryotes. Microbiology and Molecular Biology Reviews, 72, 686-727. doi:10.1128/MMBR.00011-08
- Kloss, E., Courtemanche, N. and Barrick, D. (2008) Repeat-protein folding: New insights into origins of cooperativity, stability, and topology. Archives of Biochemistry and Biophysics, 469, 83-99. doi:10.1016/j.abb.2007.08.034
- Hancock, J.M. and Simon, M. (2005) Simple sequence repeats in proteins and their significance for network evolution. Gene, 345, 113-118. doi:10.1016/j.gene.2004.11.023
- Marcotte, E.M., Pellegrini, M., Yeates, T.O. and Eisenberg, D. (1999) A census of protein repeats. Journal of Molecular Biology, 293, 151-160. doi:10.1006/jmbi.1999.3136
- Gangloff, S., Zou, H. and Rothstein, R. (1996) Gene conversion plays the major role in controlling the stability of large tandem repeats in yeast. EMBO Journal, 15, 1715- 1725.
- Vergnaud, G. and Denoeud, F. (2000) Minisatellites: Mutability and genome architecture. Genome Research, 10, 899-907. doi:10.1101/gr.10.7.899
- Armour, J.A., Povey, S., Jeremiah, S. and Jeffreys, A.J. (1990) Systematic cloning of human minisatellites from ordered array charomid libraries. Genomics, 8, 501-512. doi:10.1016/0888-7543(90)90037-U
- Denoeud, F., Vergnaud, G. and Benson, G. (2003) Predicting human minisatellite polymorphism. Genome Research, 13, 856-867. doi:10.1101/gr.574403