International Journal of Organic Chemistry
Vol. 2 No. 3 (2012) , Article ID: 22760 , 15 pages DOI:10.4236/ijoc.2012.23032
Mortar-Pestle and Microwave Assisted Regioselective Nitration of Aromatic Compounds in Presence of Certain Group V and VI Metal Salts under Solvent Free Conditions
Department of Chemistry, Osmania University, Hyderabad, India
Email: *kcrajannaou@yahoo.com
Received May 3, 2012; revised June 5, 2012; accepted June 23, 2012
Keywords: Nitration; Mortar-Pestle; Microwave-Assisted Nitration; Ammonium Molybdate; Potassium Chromate; Sodium Tungstate; Bismuth Nitrate; Sodium Bismuthate
ABSTRACT
Solvent-free Mortar-pestle (grinding) and microwave-assisted nitration reactions (MWA NR’s) underwent smoothly in the presence of group V and VI metal salts with high regio-selectivity for anilides, moderatelyand non-activated aromatic compounds. The reactions were conducted under solvent-free conditions, which afforded good to excellent yields. The observed reaction times in MW assisted conditions are in the range of only few minutes.
1. Introduction
Nitro aromatic compounds are extensively used as chemical feed stocks for a wide range of materials such as dyes, pharmaceuticals, perfumes, and plastics. Therefore, nitration of organic compounds has been a long, very active and rewarding area of research and is the subject of a large body of literature [1-4]. More specifically the nitration of benzene and toluene is sone of the most important routs to substituted aromatics in the production of chemical intermediates. The introduction of a nitro group into an aromatic ring is commonly performed in strongly acidic polar media [3-9] by means of mixed acid (a mixture of nitric acid, sulfuric acid, and water), which leads to excessive acid waste streams and added expense. Separation of the products from the acid is often a difficult and energy consuming process that habitually implies a basic aqueous work-up. Moreover, sulfuric acid is corrosive and is dangerous to transport and handle. The above mentioned disadvantages of the commercial manufacturing process currently used have led to a substantial effort to develop viable alternatives. Quite often either metal nitrates or metal nitrates supported on silica, alumina or clay [10-26] have been used as catalysts in the alternate methods of nitration to overcome the problems of classical nitration. In recent past Bismuth (III) compounds have received particular attention as low toxicity reagents and catalysts for various organic transformations [11,12].
In recent past, increasing attention has been paid to the ‘green chemistry’ processes that reduce or eliminate the use or generation of hazardous substances [13]. As a result “Atom-economy” of chemical reactions has become one of the most important key concepts of green and sustainable chemistry [14-24]. Synthetic chemists have tried and still are trying to achieve these goals by developing several valuable and distinctive techniques [25] to achieve these goals. Solvent free organic synthesis has been of great interest in recent years [26,27]. Elimination of volatile organic solvents in organic synthesis is one of the most important goals in green chemistry. Solvent free organic reactions make synthesis simpler, save energy and prevent solvent wastes, hazards and toxicity. In this part of our work we aimed at to explore solvent free nitration methods such as (a) grinding the solvent free reactants in a mortar with a pestle [28-34] and (b) conducting micro wave assisted nitration reactions [35-43].
Microwaves are a form of electromagnetic radiation. When molecules with a permanent dipole are placed in an electric field, they become aligned with that field. If the electric field oscillates, then the orientations of the molecules will also change in response to each oscillation. Most microwave ovens operate at 2.45 GHz wavelength, at which oscillations occur 4.9 × 109 times per second. Molecules subjected to this microwave radiation are extremely agitated as they align and realign themselves with the oscillating field, creating an intense internal heat that can escalate as quickly as 10˚C per second. Non-polar molecules such as toluene, carbon tetrachloride, diethyl ether and benzene are microwave inactive, while polar molecules such as DMF, acetonitrile, dichloromethane, ethanol and water are microwave active. This technique proved to be excellent in cases where traditional heating has a low efficiency because of poor heat transmission and, hence, local overheating is a major inconvenience. The most important advantage of microwave-enhanced chemistry is the reduction in the reaction times. Reactions that require hours or days of conventional heating may often be accomplished in minutes under microwave heating. Moreover, reactions are not only faster, but proceed with higher purity and, consequently, higher yields.
The proposed work is taken in three different stages 1) conventional stirring/reflux conditions in solvent phase 2) grinding the reactants in a mortar with a pestle under solvent-free conditions. 3) using microwave irradiation under solvent-free conditions to save energy.
2. Experimental Details
2.1 Materials and Methods
All chemicals used were of analytical grade. All the reagents and substrates used were of laboratory reagent grade, which were obtained from E-Merck, SDfine chemicals or Alfa Aesar. Doubly distilled water (distilled over alkaline KMnO4 and acid dichromate in an all glass apparatus) was used whenever required. Solvents were HPLC grade and used as such.
Laboratory model microwave reactor (CEM – 908010, bench mate model, 300 W equipped with temperature, pressure and microwave power control units) was used for microwave assisted reactions in this study.
2.2. Typical Experimental Procedure for Nitration of Organic Compounds under Conventional Conditions
The following procedure is a representative reaction. Phenol (0.094 ml, 1 mmol) and metal salt (394 mg, 1 mmol) were taken in chloroform (10 ml). Then 69% HNO3 (0.063 ml, 1 mmol) was added and reaction mixture was stirred at room temperature for 3hrs, after the completion of reaction as indicated by TLC, the reaction mixture was filtered off and washed with water, organic layer was separated out dried over sodium sulphate and evaporated under vacuum. The crude product was purified by chromatography using ethyl acetate: hexane (3:7) as eluent to get p-nitrophenol m.p 113˚C (lit.mp. 114˚C) yield 85% as major product.
2.3. Typical Experimental Procedure for Solvent -Free Nitration of Organic Compounds by Grinding the Reactants in a Mortar with Pestle
A mixture of the aromatic compound (1 mmol), few drops of HNO3 (1 mmol) and metal salt (1 mmol) was ground in a mortar with a pestle at room temperature, till a slurry was observed (Figure 1). Progress of the reaction was monitored with TLC. Upon completion of the reaction, the reaction mixture was treated with sodium thiosulfate; the organic layer was diluted with dichloromethane (DCM), and separated from aqueous layer. Crude product was purified by coloumn chromatography using ethyl acetate hexane as eluent. The products were identified by characteristic spectroscopic data ((Figures S.1 to S.9 in Supplementary Data).
2.4. Typical Experimental Procedure for Microwave Assisted Nitration (MWANR) of Organic Compounds
The microwave reactor used was of CEM make, which was equipped with temperature, pressure and microwave power control units. An oven-dried microwave vial was charged with a mixture containing aromatic compound, metal nitrate and few drops of nitric acid and silica gel slurry, and irradiated in a microwave (power input 140 W) at 150˚C for few minutes. After completion of the reaction, as ascertained by TLC, the reaction mixture was treated with sodium thiosulfate; the organic layer was diluted with dichloromethane (DCM), and separated from aqueous layer. Crude product mixture was purified with ethyl acetate DCM mixture. The purity was checked with TLC. The products were identified by characteristic spectroscopic data (Figures S.1 to S.9 in Supplementary Data)
3. Results & Discussion
Data presented in Tables 1 to 5 represent certain group - V metal salts (bismuth nitrate (BN), sodium bismuthate (SB)) and certain group - VI B metal salts such as potassium chromate (PCR), ammonium molybdate (AMB) and sodium tungstate (STG) ) which are used as catalysts to onset nitration of non-active and moderately active aromatic

Figure 1. Grinding the reactants in a mortar with a pestle under solvent-free conditions.
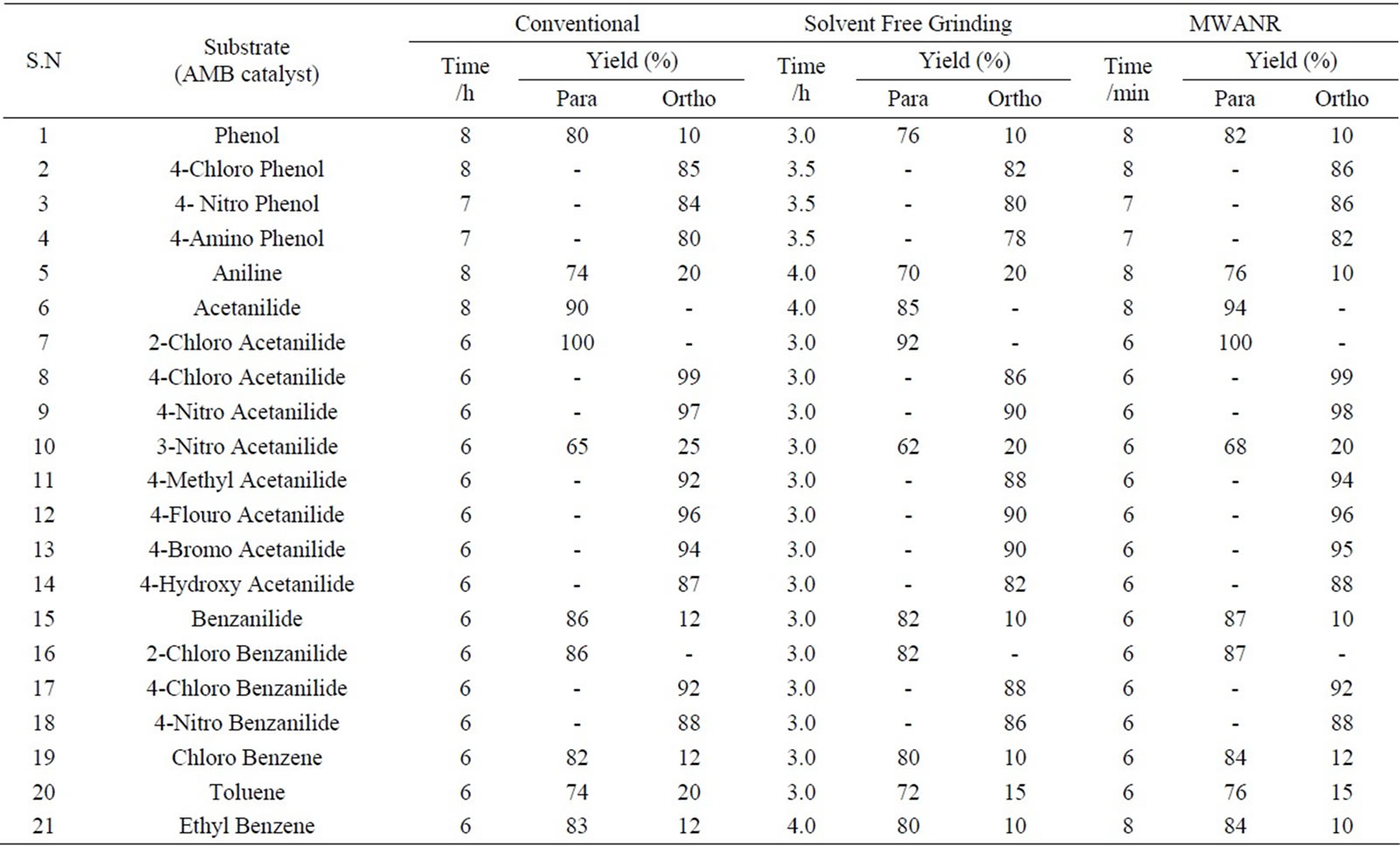
Table 1. Microwave assisted mmonium molybdate mediated regio selective nitration of anilides, non-activated and moderately activated organic compounds under mild acid conditions.
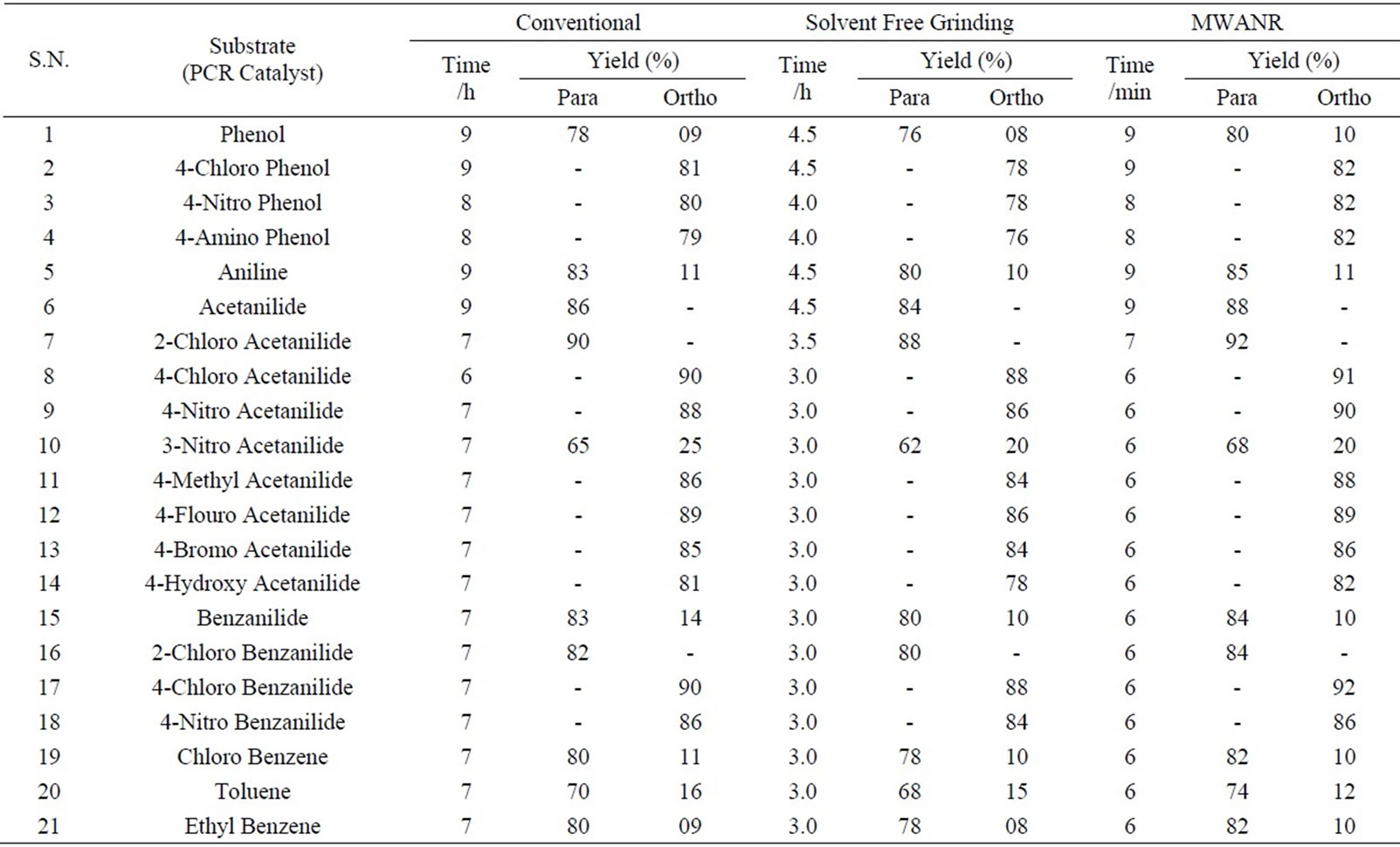
Table 2. Microwave assisted potassium chromate catalyzed regio selective nitration of anilides, non-activated and moderately activated organic compounds under mild acid conditions.
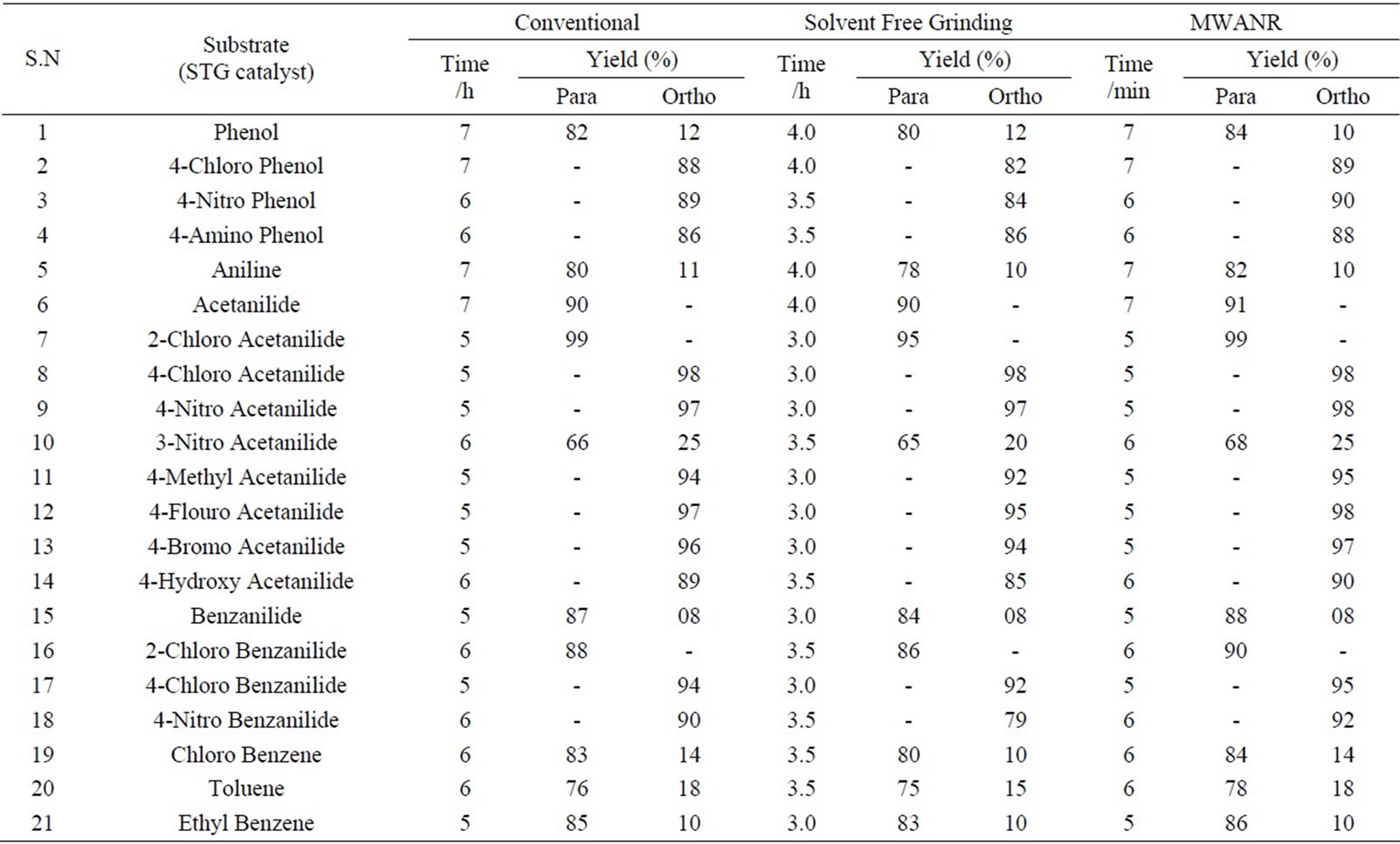
Table 3. Microwave assisted sodium tungstate catalyzed regio selective nitration of anilides, non-activated and moderately activated organic compounds under mild acid conditions.
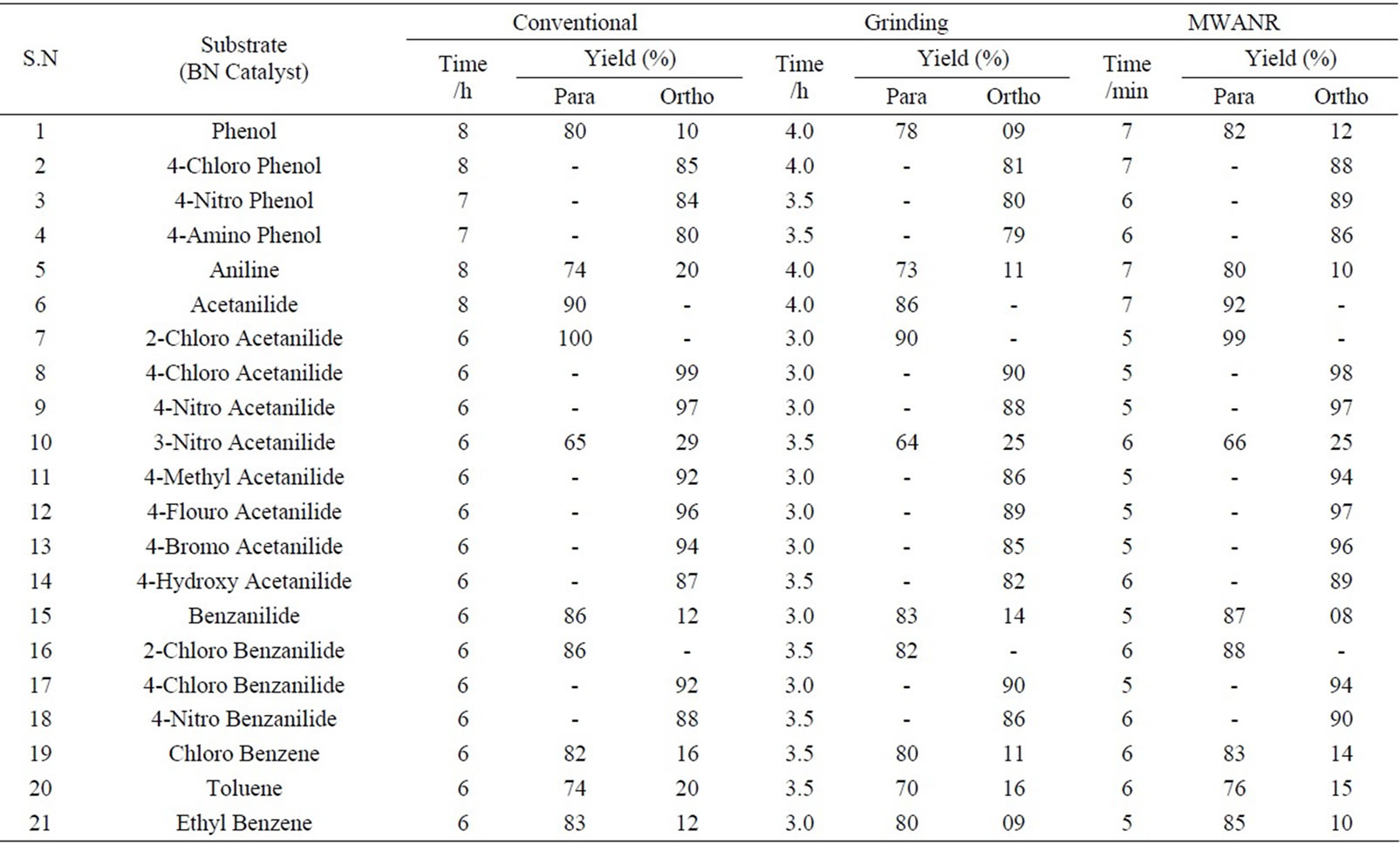
Table 4. Microwave assisted bismuth nitrate catalyzed regio selective nitration of anilides, non-activated and moderately activated organic compounds under mild acid conditions.
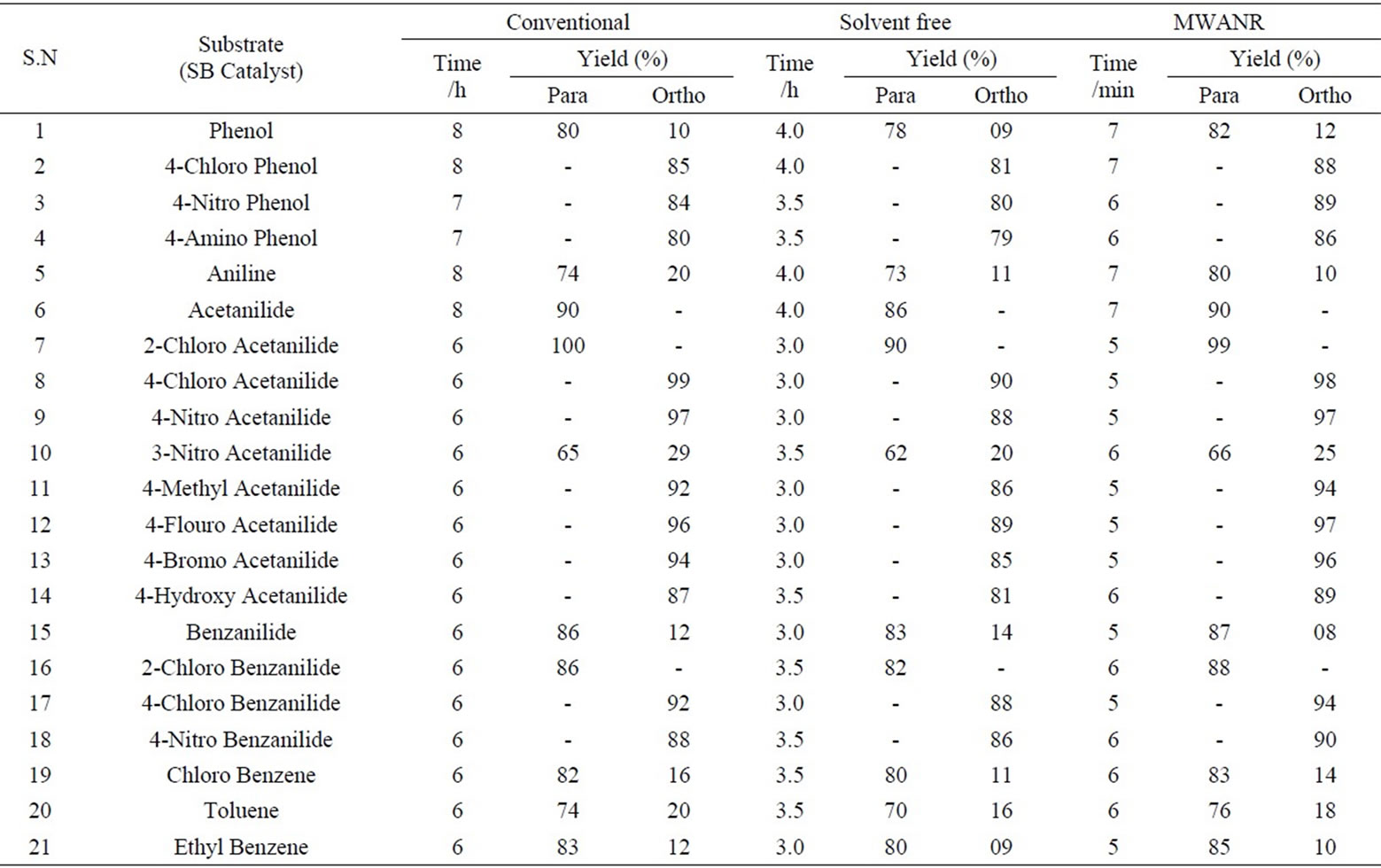
Table 5. Microwave assisted sodium bismuthate catalyzed regio selective nitration of anilides, non-activated and moderately activated organic compounds under mild acid conditions.
compounds, under conventional and non-conventional conditions. Solvent-free grinding and microwave assisted methods were chosen as non-conventional techniques. Traditional nitration reactions underwent smoothly with moderate to long reaction times (6 to 8 hours) with good yields with good regioselectivity (Scheme 1).
However, the active aromatic compounds such as carbonyl compounds underwent within hour affording high yields of the corresponding mono nitro derivatives (Tables 6 to 10) with high regioselectivity (Scheme 1). The reactions were clean, no attack being observed on the alkyl portion of the ketones. In marked contrast to ordinary nitration using mixed acid, which predominantly lead to metasubstitutions. In the absence of metal salts, the nitration did not proceed.
Solid state reaction occurred more efficiently and more selectively than the corresponding solution phase reactions, since molecules in the crystal are arranged tightly and regularly [34]. In present work grinding technique appears to be superior since it is eco-friendly, high yielding, requires no special apparatus, non-hazardous, simple and convenient. Rate accelerations could be explained due to the conversion of mechanical energy (kinetic energy exerted due to grinding) into heat energy, which becomes driving force for better activation of molecules. The kinetic energy supplied during grinding can have several effects on a crystalline solid [28-34] including: heating, reduction of particle size (with concomitant increase in surface area and the generation of fresh surfaces), formation of defects and dislocations in crystal lattices, local melting and even phase changes to alternative polymorphs. Collisions between crystals during grinding can also lead to local deformations and potentially melting. Importantly, grinding also provides mass transfer, i.e. it is a sort of ‘stirring’.
The dramatic acceleration and increased purity and yields of microwave assisted reactions make them attractive to the increased demands in industry and, in particular, for combinatorial drug discovery. In addition to being energy efficient, the possibility of employing milder and less toxic reagents and solvents, or even solvent-free
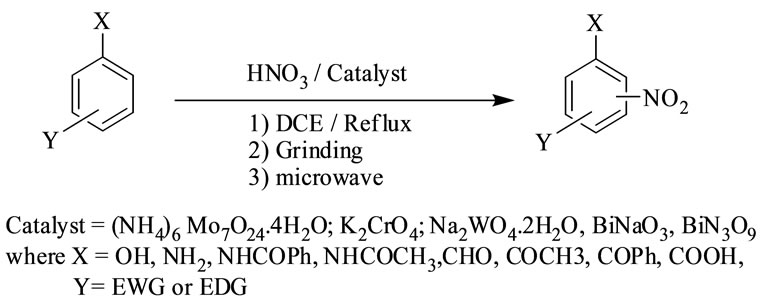
Scheme 1. Nitration of organic compounds catalysed by group V and VI metal salts under solvent free conditions.
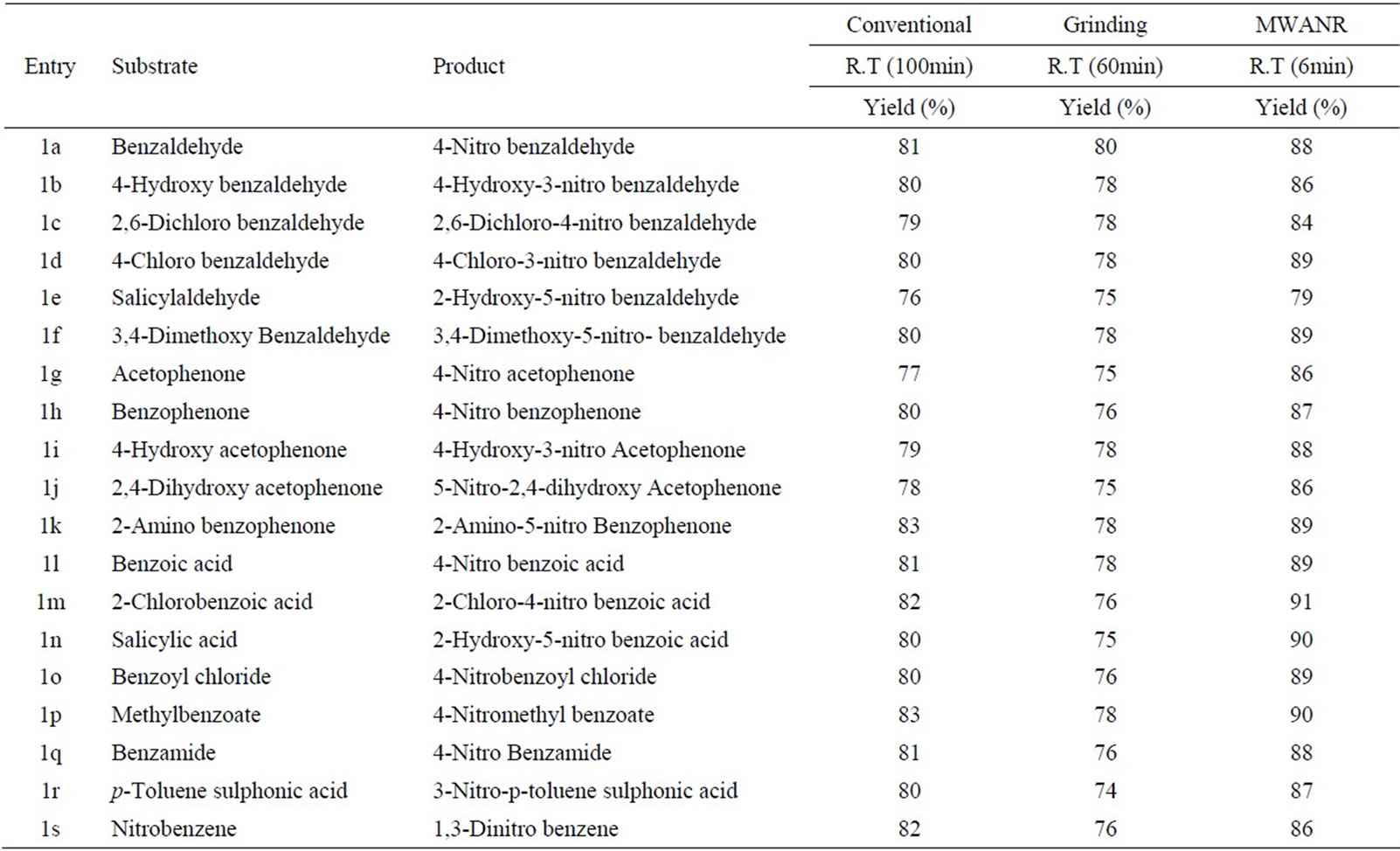
Table 6. Microwave Assisted Potassium Chromate catalysed Nitration of Carbonyl and Related Compounds under mild acid conditions.
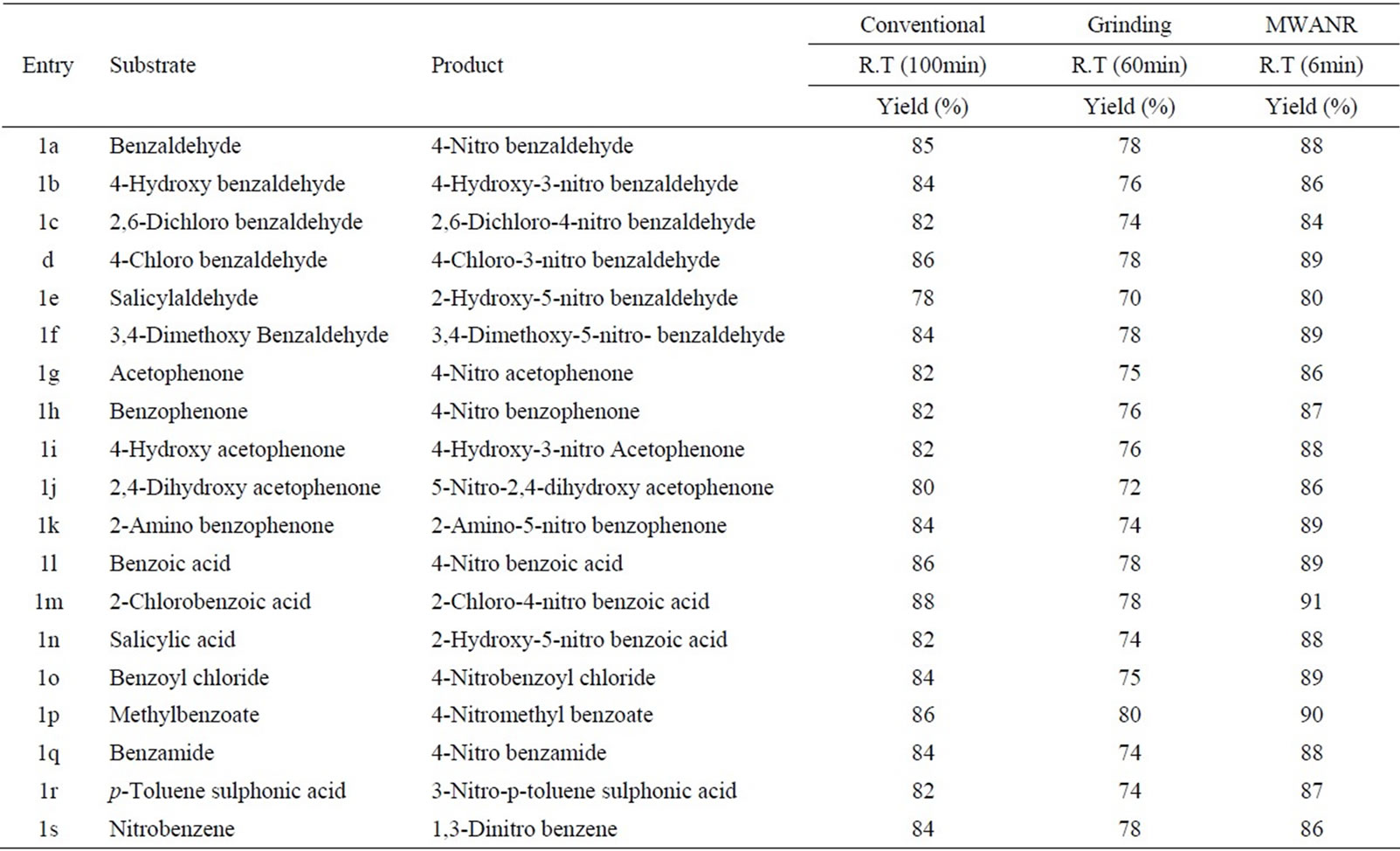
Table 7. Microwave assisted ammonium molybdate catalysed nitration of carbonyl and related compounds under mild acid conditions.
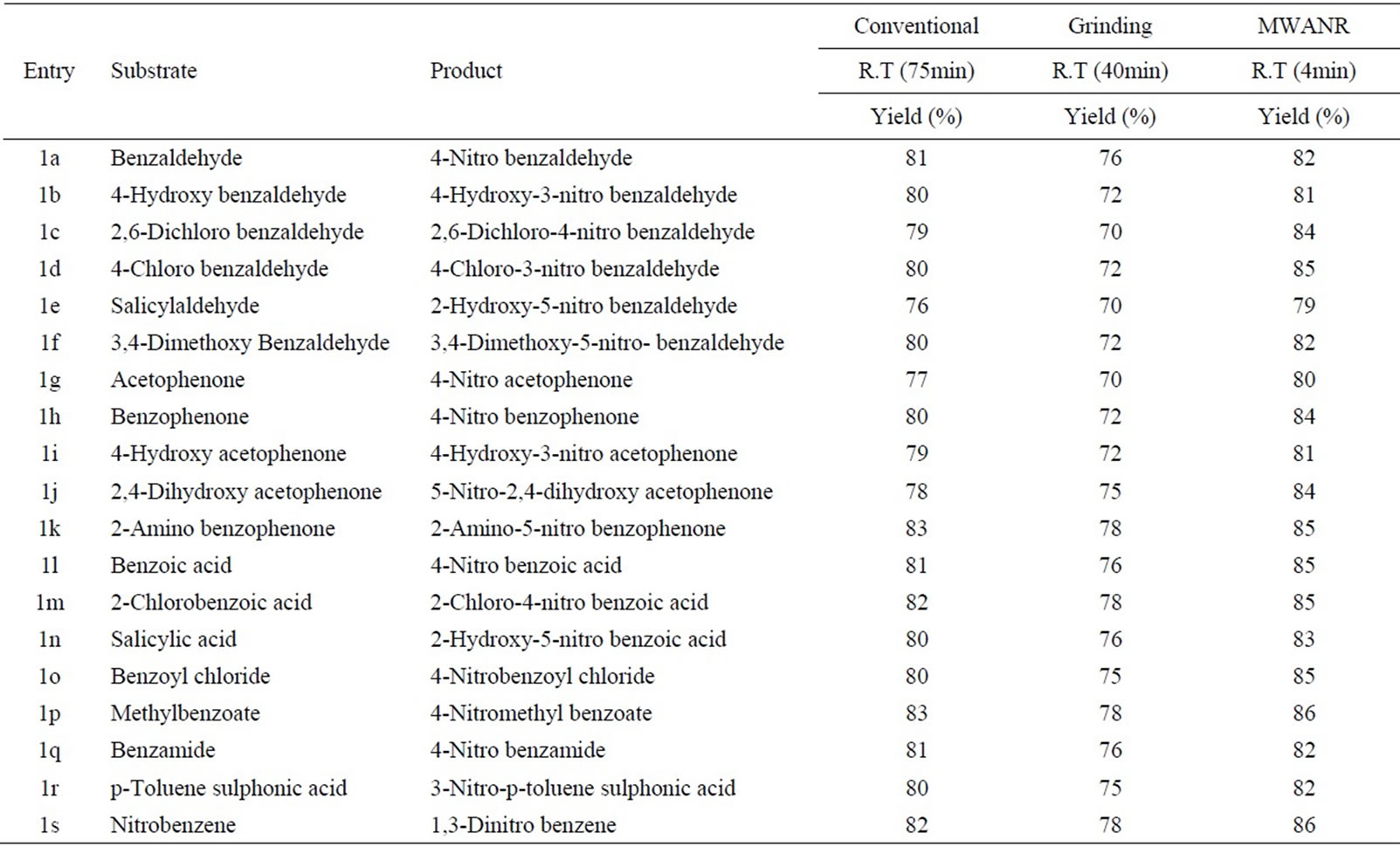
Table 8. Microwave assisted sodium tungstate catalysed nitration of carbonyl and related compounds under mild acid conditions.
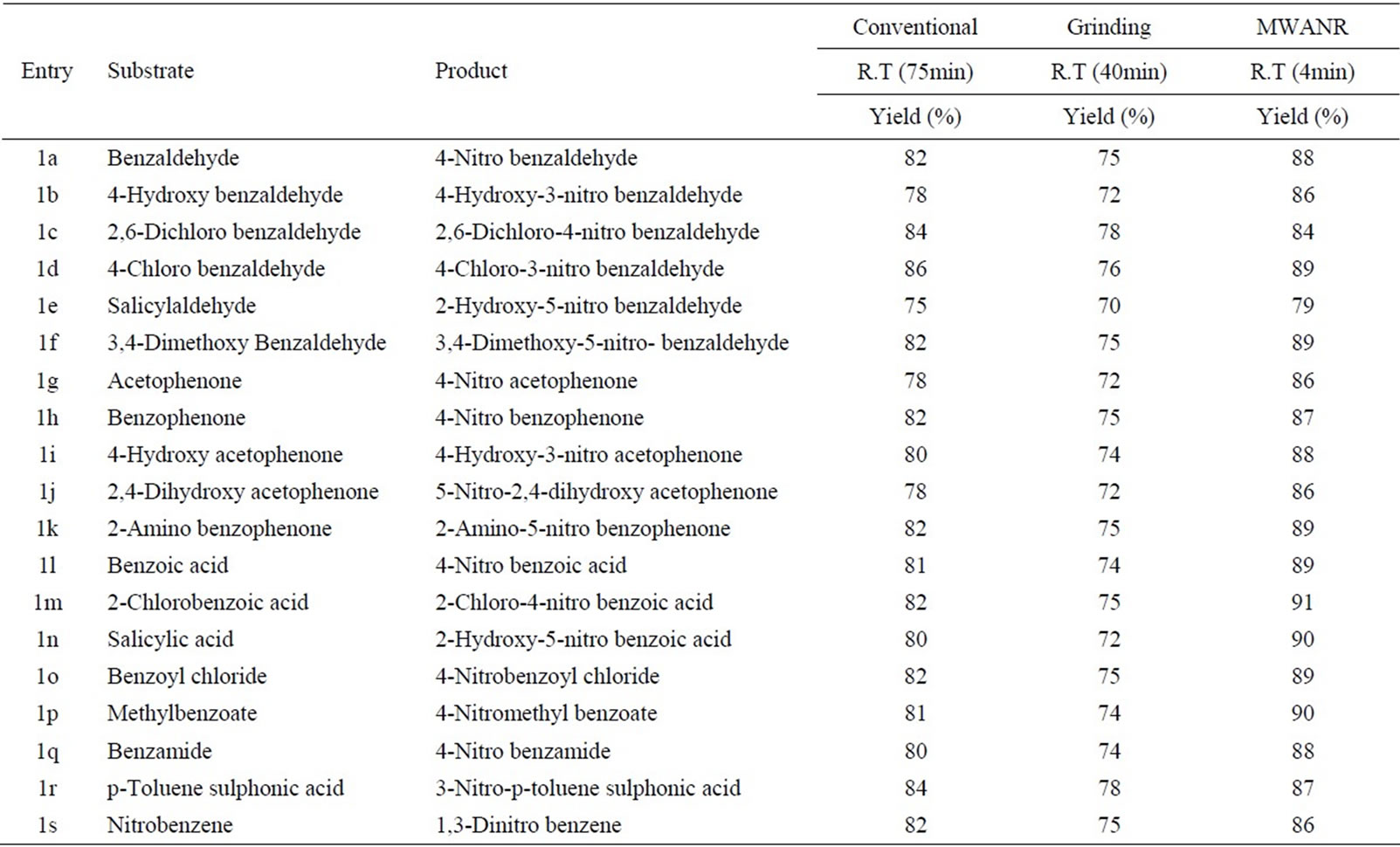
Table 9. Microwave assisted sodium bismuthate catalysed nitration of carbonyl and related compounds under mild acid conditions.
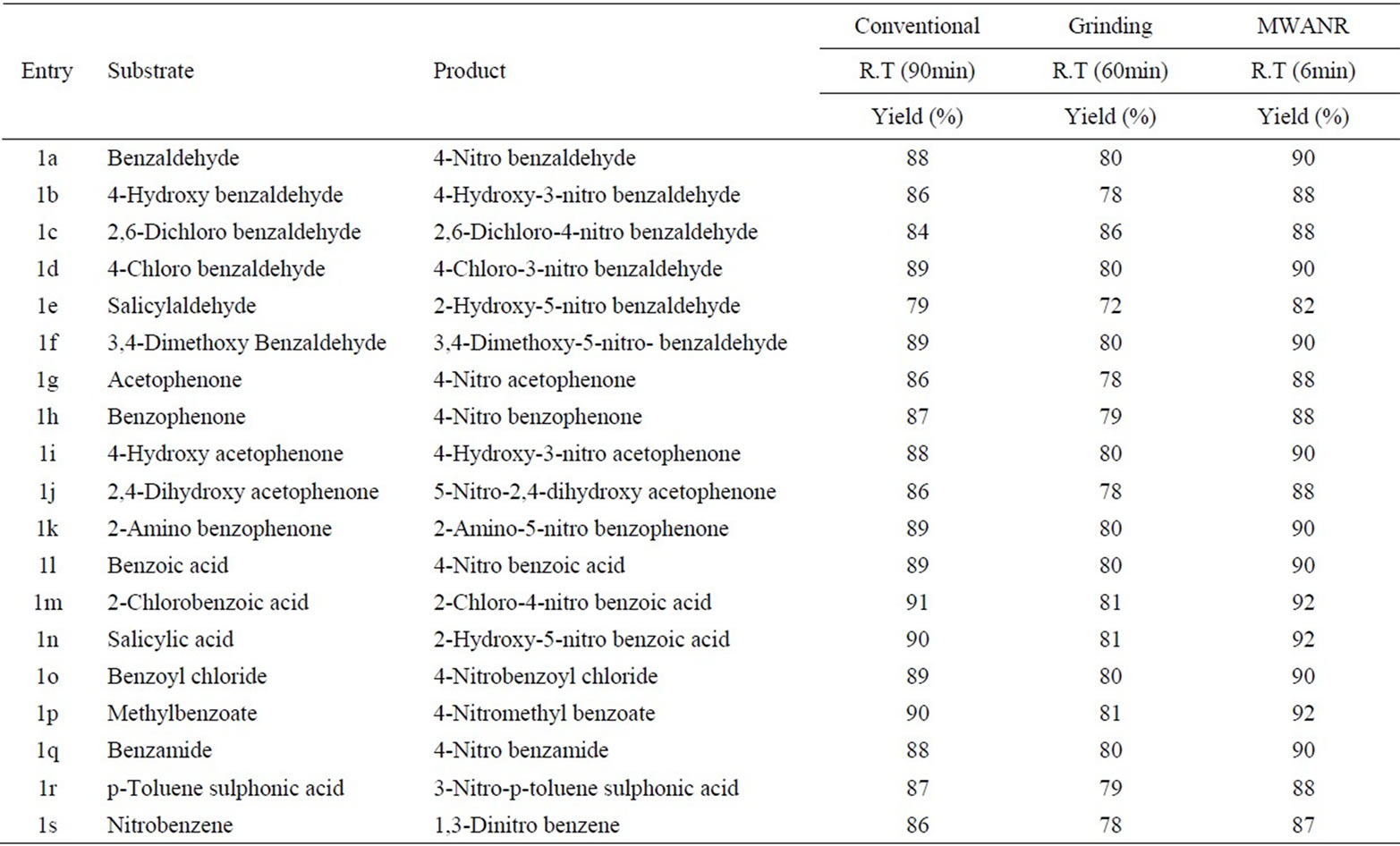
Table 10. Microwave assisted bismuth nitrate catalysed nitration of carbonyl and related compounds under mild acid conditions.
systems, offers a further advantage of this heating technology. In order to check for a possible specific (not purely thermal) microwave effect, CEM model bench mate microwave oven was used [44]. Under conventional conditions an increase in temperature increases only fraction of activated molecules. At any given time temperature on the surface of the reaction vessel is greater than the internal temperature, and heat energy is transferred to the reaction mixture via thermal conduction. However, in MW assisted reactions microwave radiation is directly transferred to reactant species. Reaction mixture absorbs microwave energy, which probably causes super heating followed by the formation of bulk activation molecules (Figures 2). Regarding the goal of a general interpretation of specific microwave effects, we can assume that these will be favorable if the polarity of the transition state is increased during the reaction (microwave materials interactions are enhanced with polarity) [45]. This should therefore be the case for reactions in which the transition state (TS) is more polar than the ground state (GS) (Figure 3) [46,47].
4. Conclusion
In conclusion, we have demonstrated that mortar-pestle (grinding) and micro wave-assisted nitration reactions (MWANR’s) underwent smoothly in the presence of
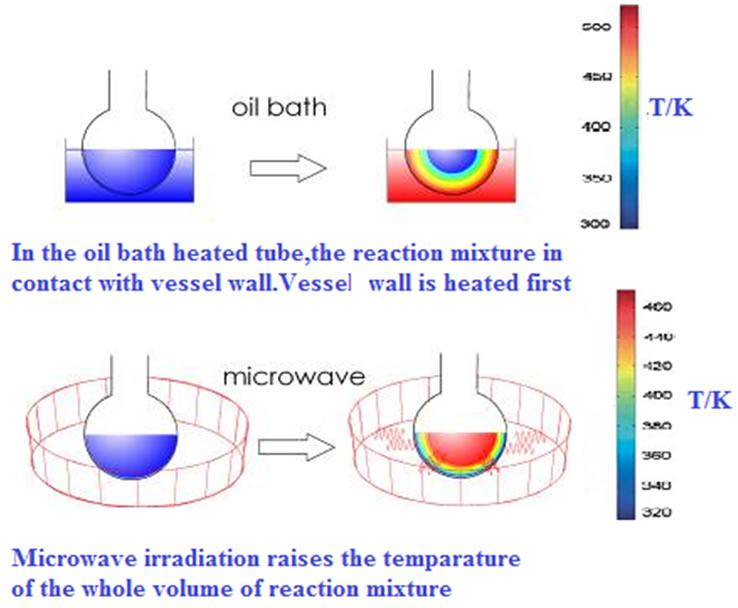
Figure 2. Microwave Assisted Nitration (MWANR) of Organic compounds.
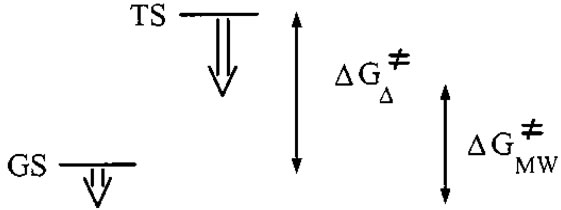
Figure 3. Relative stabilization of transition state (TS) and ground state (GS) by dipole-dipole interactions with electromagnetic field if TS is more polar than GS.
group V and VI metal salts for the first time. These methods have several advantages over existing methods such as region-selectivity, high yields, simple procedure, and short reaction times. It is noteworthy to mention here that if the ortho position is engaged, p-nitro derivatives are obtained while o-nitro derivatives are obtained when para position is engaged. In case of MWANR of aromatic carbonyl and related compounds the effect of microwaves is extremely high. The observed reaction times are in the range of 3 - 5 minutes.
5. Electronic Supplementary Material
Figures S.1 to S.9 in Supplementary Data indicate certain spectroscopic results of nitration products.
REFERENCES
- G. Booth, “Nitro Compounds, Aromatic,” Ullmann’s Encyclopedia of Industrial Chemistry, Wiley-VCH, Weinheim, 2005.
- G. A. Olah, R. Malhorta and S. C. Narang, “Nitration: Methods and Mechanisms,” VCH Publishers, New York, 1989.
- H. Zollinger, “Color Chemistry: Properties and Applications of Organic Dyes,” 2nd Edition, John Wiley, New York, 1991.
- R. Meyer, J. Kholar and A. Homburg, “Explosives,” 5th Edition, John Wiley, New York, 2002.
- M. B. Smith and J. March, “March’s Advanced Organic Chemistry,” 5th Edition, John Wiley, New York, 2001.
- S. P. Dagade, S. B. Waghmode, V. S. Kadam and M. K. Dongare, “Vapor Phase Nitration of Toluene Using Dilute Nitric Acid and Molecular Modeling Studies over Beta Zeolite,” Applied Catalysis A: General, Vol. 226, No. 13, 2002, pp. 49-61. doi:10.1016/S0926-860X(01)00882-1
- T. Esakkidurai and K. Pitchumani, “Zeolite-Mediated Regioselective Nitration of Phenol in Solid State,” Journal of Molecular Catalysis A: Chemical, Vol. 185, No. 1-2, 2002, pp.305-309. doi:10.1016/S1381-1169(02)00135-8
- M. A. Zolfigol, E. Ghaemi and E. Madrakian, “Trichloroisocyanuric Acid/NaNO2 as a Novel Heterogeneous System for the Selective Mononitration of Phenols under Mild Conditions,” Synlett, No. 2, 2003, pp.191-194. doi:10.1002/chin.200319081
- H. A. Muathen, “Selective Nitration of Aromatic Compounds with Bismuth Subnitrate and Thionyl Chloride,” Molecules, Vol. 8, No. 7, 2003, pp. 593-598. doi:10.3390/80700593
- N. M. Leonard, L. C. Wieland and R. S. Mohan, “Applications of Bismuth(III) Compounds in Organic Synthsis,” Tetrahedron, Vol. 58, No. 42, 2002, pp. 8373-8397. doi:10.1016/S0040-4020(02)01000-1
- H. Suzuki, T. Ikegami and Y. Matano, “Bismuth in Organic Transformations,” Synthesis, No. 3, 1997, pp. 249- 267. doi: 0.1055/s-1997-1194
- J. H. Ridd, “Some Unconventional Pathways in Aromatic Nitration,” Acta Chemica Scandinavica, Vol. 52, No. 1, 1998, pp. 11-22. doi:10.3891/acta.chem.scand.52-0011
- P. T. Anastas and J. C. Warner, “Green Chemistry: Theory and Practice,” Oxford University Press, New York, 1998.
- P. T. Anastas and T. C. Williamson, “Green Chemistry: Designing Chemistry for the Environment,” American Chemical Society, Washington, DC, 1996.
- P. T. Anastas and L. G. Heine, “Green Chemical Synthesis and Processes,” American Chemical Society, Washington, DC, 2000.
- S. K. Ritter, Chem. Eng. News 2001, pp 27-34.
- P.T. Anastas, M. M. Kirchhoff, “Origin, Current Status, and Future Challenges of Green Chemistry,” Accounts of Chemical Research, Vol. 35, No. 9, 2002, pp. 686- 694. doi:10.1021/ar010065m
- M. Lancaster, “Green Chemistry: An Introductory Text,” RSC, Cambridge, 2002.
- A. Yamamoto, “Toward Development of Environmentally Benign Processes Catalyzed by Transition-Metal Complexes,” Pure and Applied Chemistry, Vol. 74, No. 1, 2002, pp. 1-6. doi:10.1351/pac200274010001
- M. Eissen and J. O. Metzger, “Environmental Performance Metrics for Daily Use in Synthetic Chemistry,” Chemistry—A European Journal, Vol. 8, No. 16, 2002, pp. 3580-3585. doi:10.1002/1521-3765(20020816
- B. M Trost, “The Atom Economy: A Search for Synthetic Efficiency,” Science, Vol. 254, No. 5037, 1991, pp. 1471- 1477. doi:10.1126/science.1962206
- B. M. Trost, “Atom Economy—A Challenge for Organic Synthesis: Homogeneous Catalysis Leads the Way,” Angewandte Chemie International Edition in English, Vol. 34, No. 3, 1995, pp. 259-281. doi:10.1002/anie.199502591
- B. M. Trost, “On Inventing Reactions for Atom Economy,” Accounts of Chemical Research, Vol. 35, No. 9, 2002, pp. 695-705. doi:10.1021/ar010068z
- D. M. P. Mingos and I. P. Beletskaya, Eds. “Atom Efficient Organic Synthesis,” Journal of Organometallic Chemistry, Vol. 23, 2004, pp. 689-697.
- F. Alonso, I. P. Beletskaya and Miguel Yusa, “Non-Conventional Methodologies for Transition-Metal Catalysed Carbon–Carbon Coupling: A Critical Overview. Part 1: The Heck Reaction,” Tetrahedron, Vol. 61, No. 50, 2005, pp. 11771-11835. doi:10.1016/j.tet.2005.08.054
- D. C. Dittmer, “‘No-Solvent’ Organic Synthesis,” Chemistry & Industry, No. 19, 1997, pp. 779-784.
- A. Kumar and S. Sharma, “A Grinding-Induced Catalystand Solvent-Free Synthesis of Highly Functionalized 1,4- Dihydropyridines via a Domino Multicomponent Reaction,” Green Chemistry, Vol. 13, No. 8, 2011, pp. 2017- 2020. doi:10.1039/C1GC15223H
- K. Tanaka and F. Toda, “Solvent-Free Organic Synthesis,” Chemical Reviews, Vol. 100, No. 3, 2000, pp. 1025- 1074.
- A. Loupy, “Solvent-Free Reactions,” Modern Solvents in Organic Synthesis, Vol. 206, 1999, pp. 153-207. doi:10.1007/3-540-48664-X_7
- R. A. Sheldon, “Green Solvents for Sustainable Organic Synthesis: State of the Art,” Green Chemistry, Vol. 7, No. 5, 2005, pp. 267-278. doi:10.1039/B418069K
- C. Suryanarayana, “Mechanical Alloying and Milling” Progress in Materials Science, Vol. 46, No. 1-2, 2001, pp. 1-184.
- R. Janot and D. Guérard, “Ball-Milling in Liquid Media: Applications to the Preparation of Anodic Materials for Lithium-Ion Batteries,” Progress in Materials Science, Vol. 50, No. 1, 2005, pp. 1-92. doi:10.1016/S0079-6425(03)00050-1
- A. L. Garay, A. Pichon and S. L. James., “Solvent-Free Synthesis of Metal Complex,” Chemical Society Reviews, Vol. 36, No. 6, 2007, pp. 846-855. doi:10.1039/b600363j
- A. Orita, L. S. Jiang, T. Nakano, N. Ma and J. Otera, “Solventless Reaction Dramatically Accelerates Supramolecular Self-Assembly,” Chemical Communications, No. 13, 2002, pp.1362. doi:10.1039/b203651g
- P. Lidstrom, J. Tierney, B. Wathey and J. Westman, “Microwave Assisted Organic Synthesis—A Review,” Tetrahedron, Vol. 57, No. 45, 2001, pp. 9225-9283. doi.10.1016/S0040-4020(01)00906-1
- C. O. Kappe and D. Dallinger, “The Impact of Microwave Synthesis on Drug Discovery,” Nature Reviews Drug Discovery, Vol. 5, No. 1, 2006, pp. 51-63. doi:10.1038/nrd1926
- A. K. Nagariya, A. K. Meena, K. Kiran, A. K. Yadav, U. S. Niranjan, A. K. Pathak, B. Singh and M. M. Rao, “Microwave Assisted Organic Reaction as New Tool in Organic Synthesis,” Journal of pharmacy Research, Vol. 3, 2010, pp.575-580.
- F. Toda, “Solid State Organic Chemistry: Efficient Reactions, Remarkable Yields, and Stereoselectivity,” Accounts of Chemical Research, Vol. 28, No. 12, 1995, pp. 480-486. doi:10.1021/ar00060a003
- R. S. Varma, “Clay and Clay-Supported Reagents in Organic Synthesis,” Tetrahedron, Vol. 58, No. 7, 2002, pp 1235-1255. doi:10.1016/S0040-4020(01)01216-9,
- M. Kidwai, R. Venkataraman and B. Dave. “Solventless Synthesis of Thiohydantoins over K2CO3,” Green Chemistry, Vol. 3, No. 6, 2001, pp. 278-279. doi:10.1039/B106034C
- C. O. Kappe., “Controlled Microwave Heating in Modern Organic Synthesis,” Angewandte Chemie International Edition, Vol. 43, No. 46, 2004, pp. 6250-6284. doi:10.1002/anie.200400655
- C. O. Kappe and A. Stadler, “Microwaves in Organic and Medicinal Chemistry,” Wiley-VCH, Weinheim, 2005.
- A. Loupy, “Microwaves in Organic Synthesis,” WileyVCH, Weinheim, 2005.
- B. Botta, G. Delle Monache, G. Zappia, et al., “Synthesis and Interaction with Copper(II) Cations of Cyanoand Aminoresorcinarenas,” The Journal of Organic Chemistry, Vol. 67, No. 4, 2002, pp. 1178-1183. doi:10.1021/jo010844g
- R. N. Gedye, F. E. Smith and K. C. Westaway, “The Rapid Synthesis of Organic Compounds in Microwave Ovens,” Canadian Journal of Chemistry, Vol. 66, No. 1, 1988, pp. 17-26. doi:10.1139/v88-003
- A. Loupy, L. Perreux, M. Liagre, K. Burle and M. Moneuse, “Reactivity and Selectivity under Microwaves in Organic Chemistry. Relation with Medium Effects and Reaction Mechanisms,” Pure and Applied Chemistry, Vol. 73, No. 1, 2001, pp. 161-166. doi:10.1351/pac200173010161
- L. Perreux and A. Loupy, “A Tentative Rationalization of Microwave Effects in Organic Synthesis According to the Reaction Medium, and Mechanistic Considerations,” Tetrahedron, Vol. 57, No. 45, 2001, pp. 9199-9223. doi:10.1016/S0040-4020(01)00905-X.
Supplementary Data
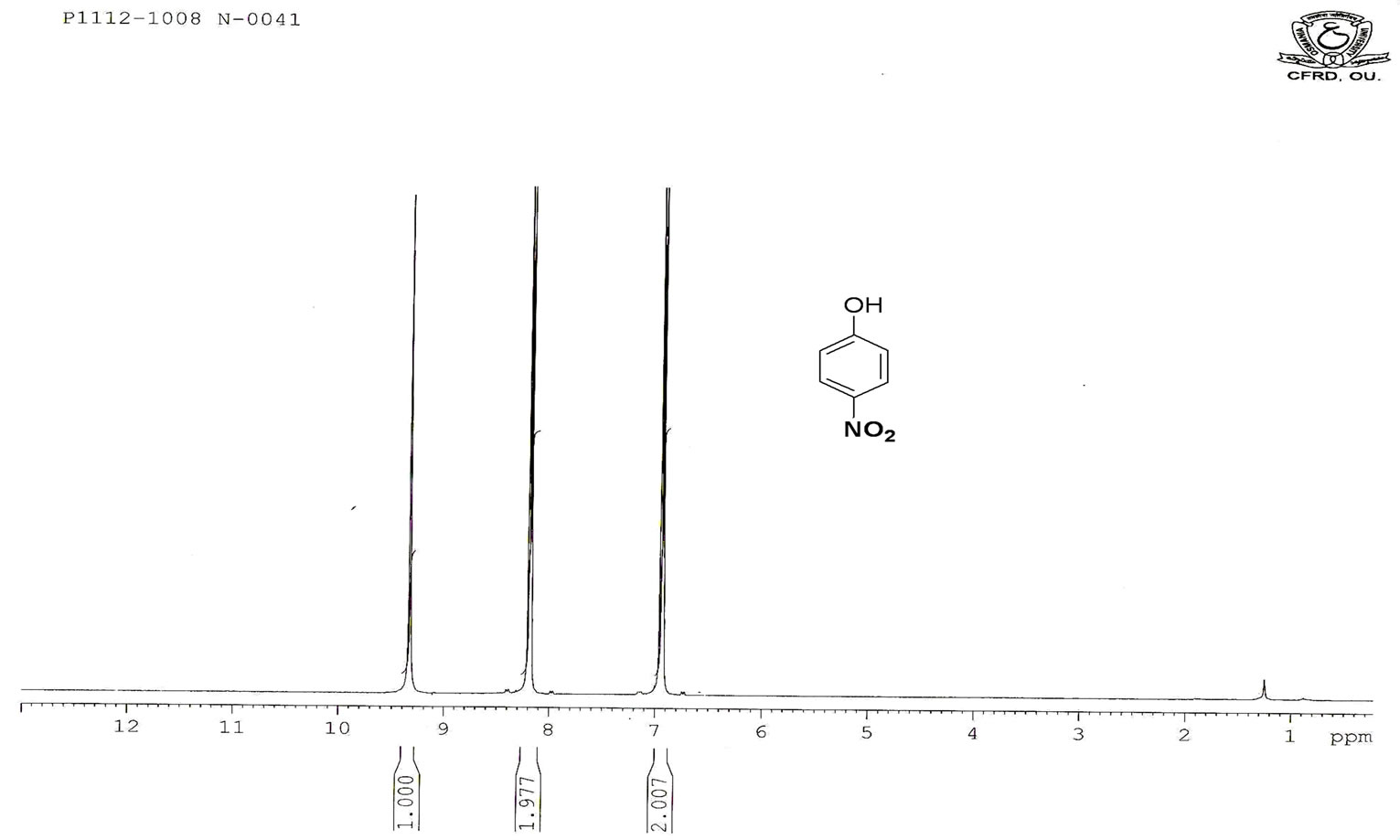
Figure S.1. HNMR Spectrum of 4-nitro phenol.
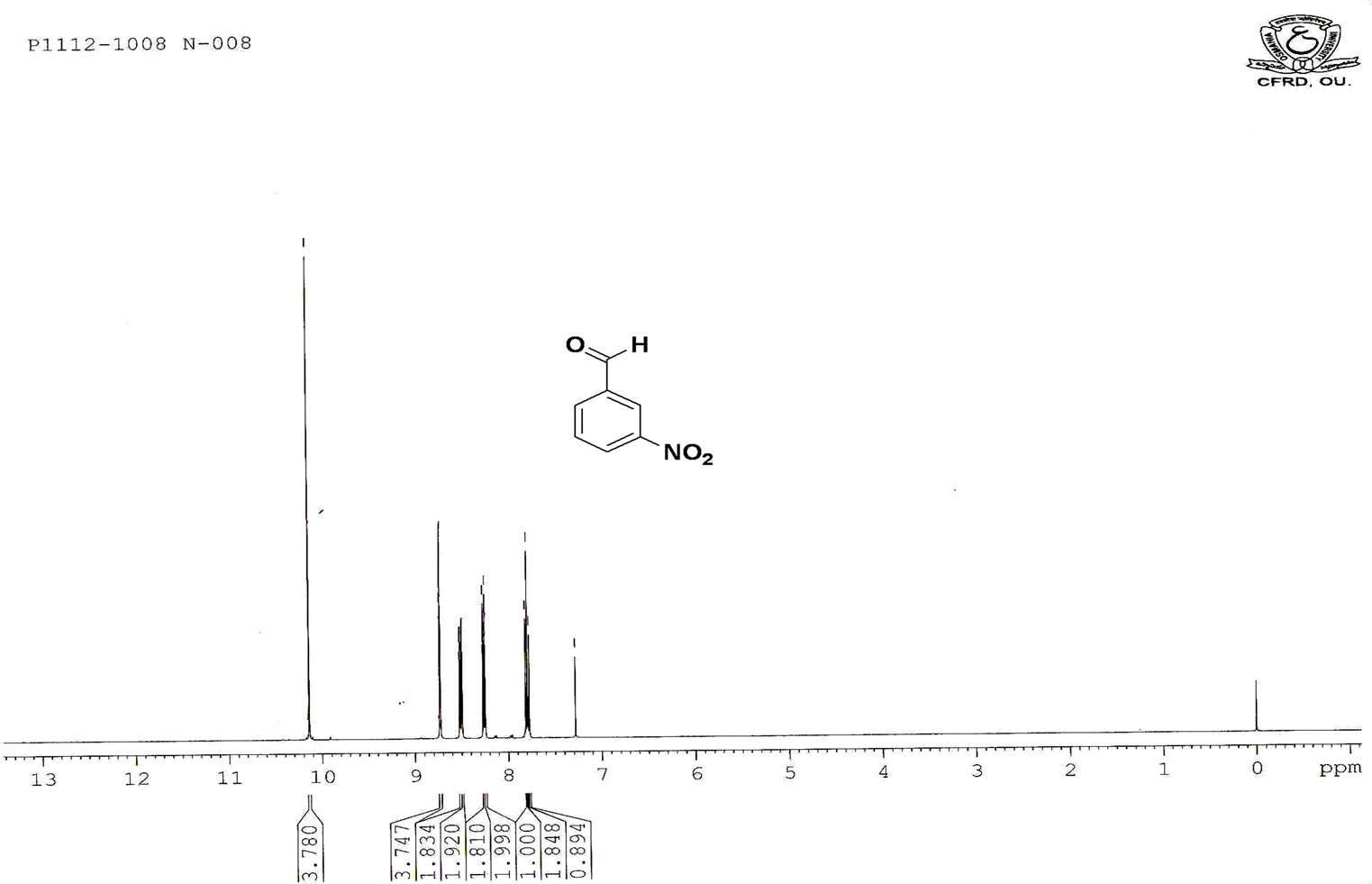
Figure S.2. HNMR Spectrum of 3-nitro benzaldehyde.
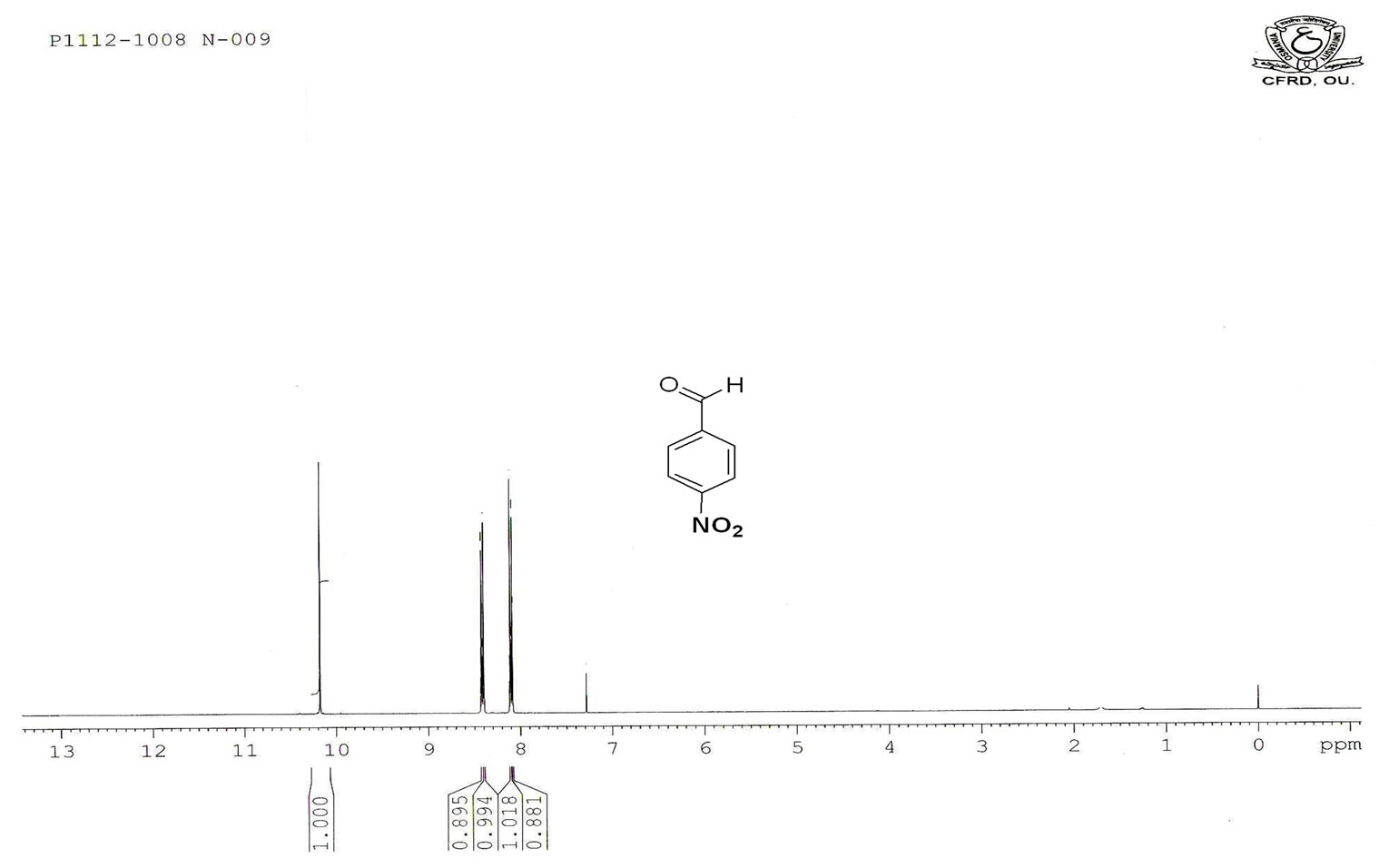
Figure S.3. HNMR Spectrum of 4-nitro benzaldehyde.
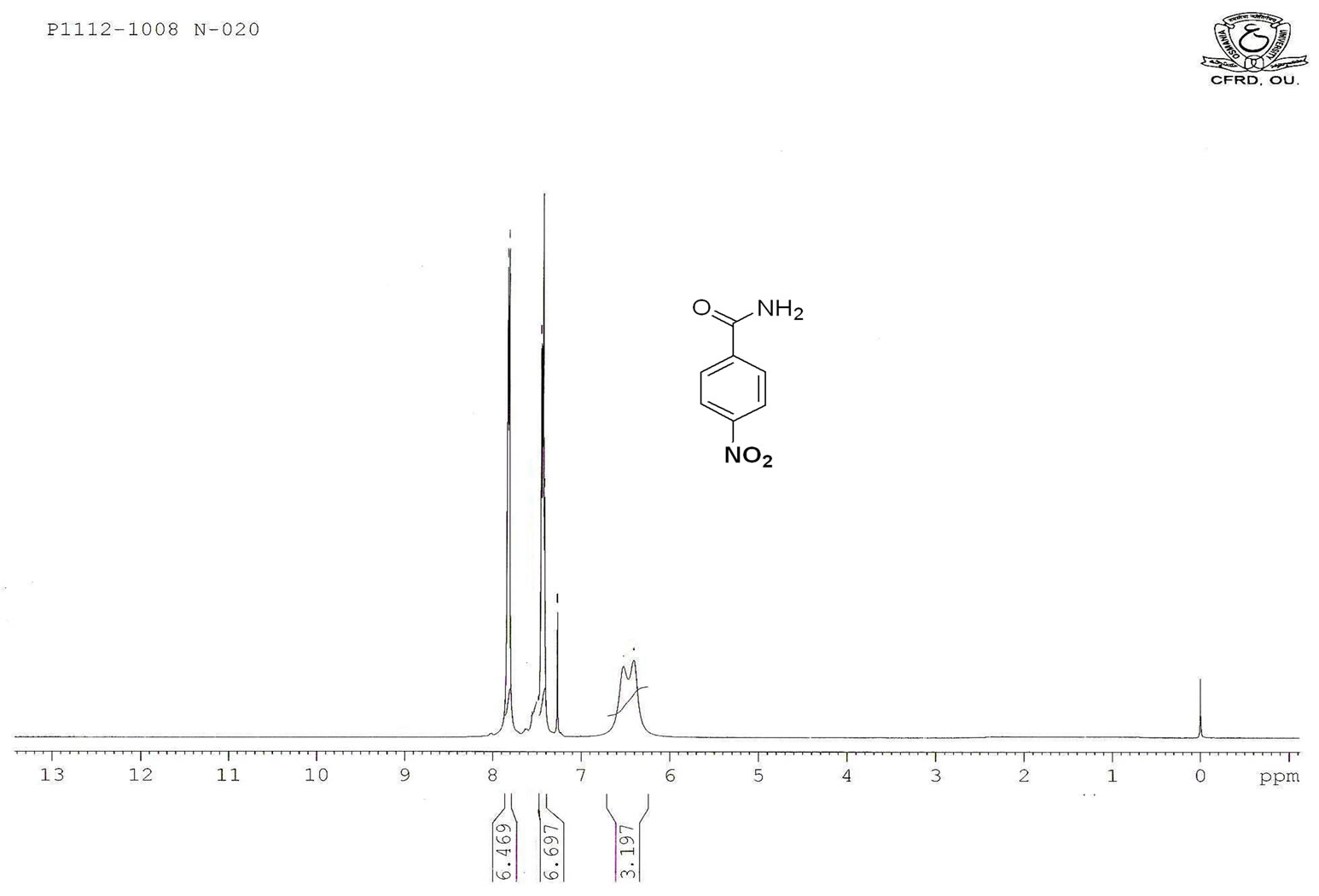
Figure S.4. HNMR Spectrum of 4-nitro benzamide.
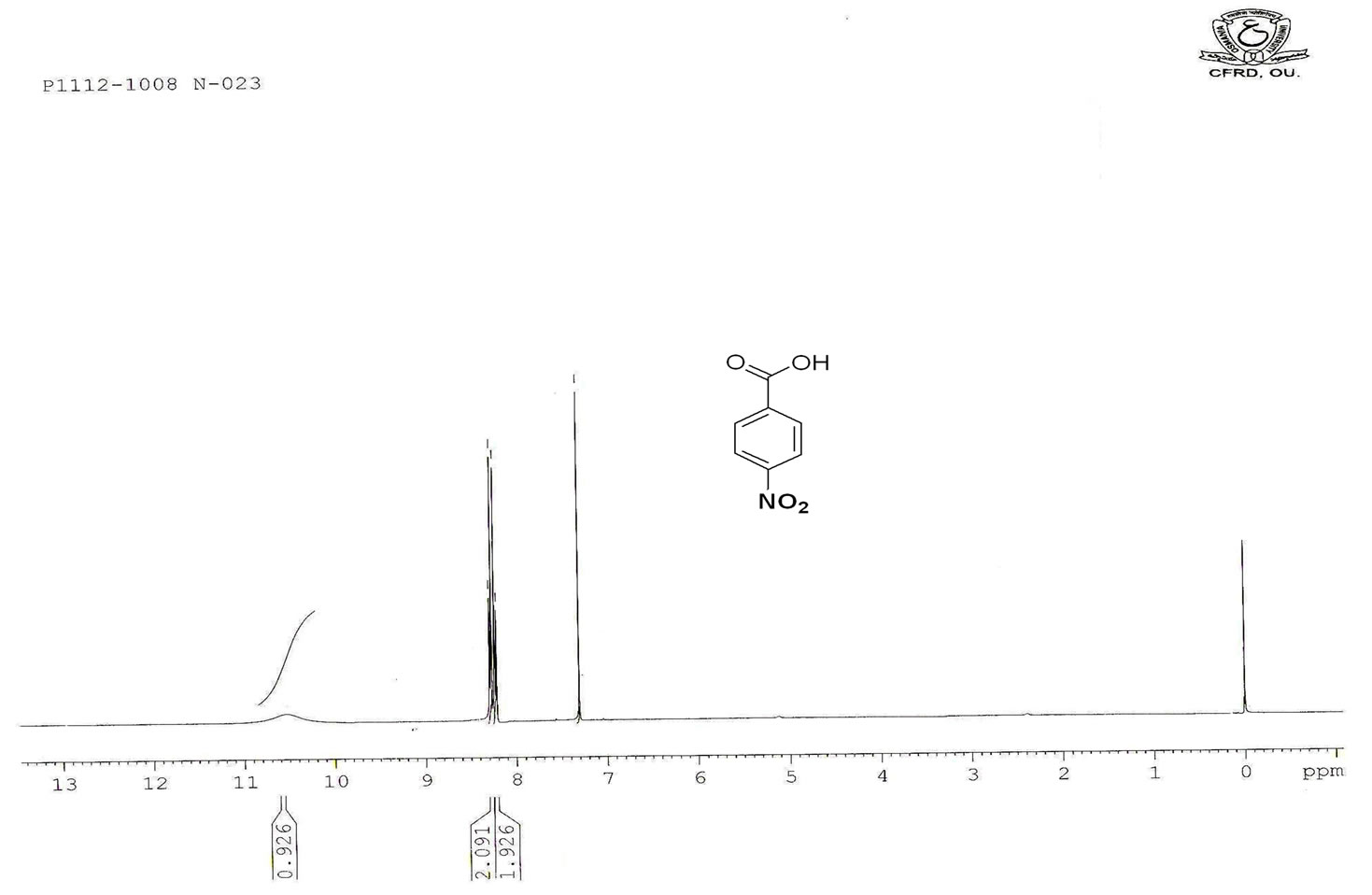
Figure S.5. HNMR Spectrum of 4-nitro benzoic acid.
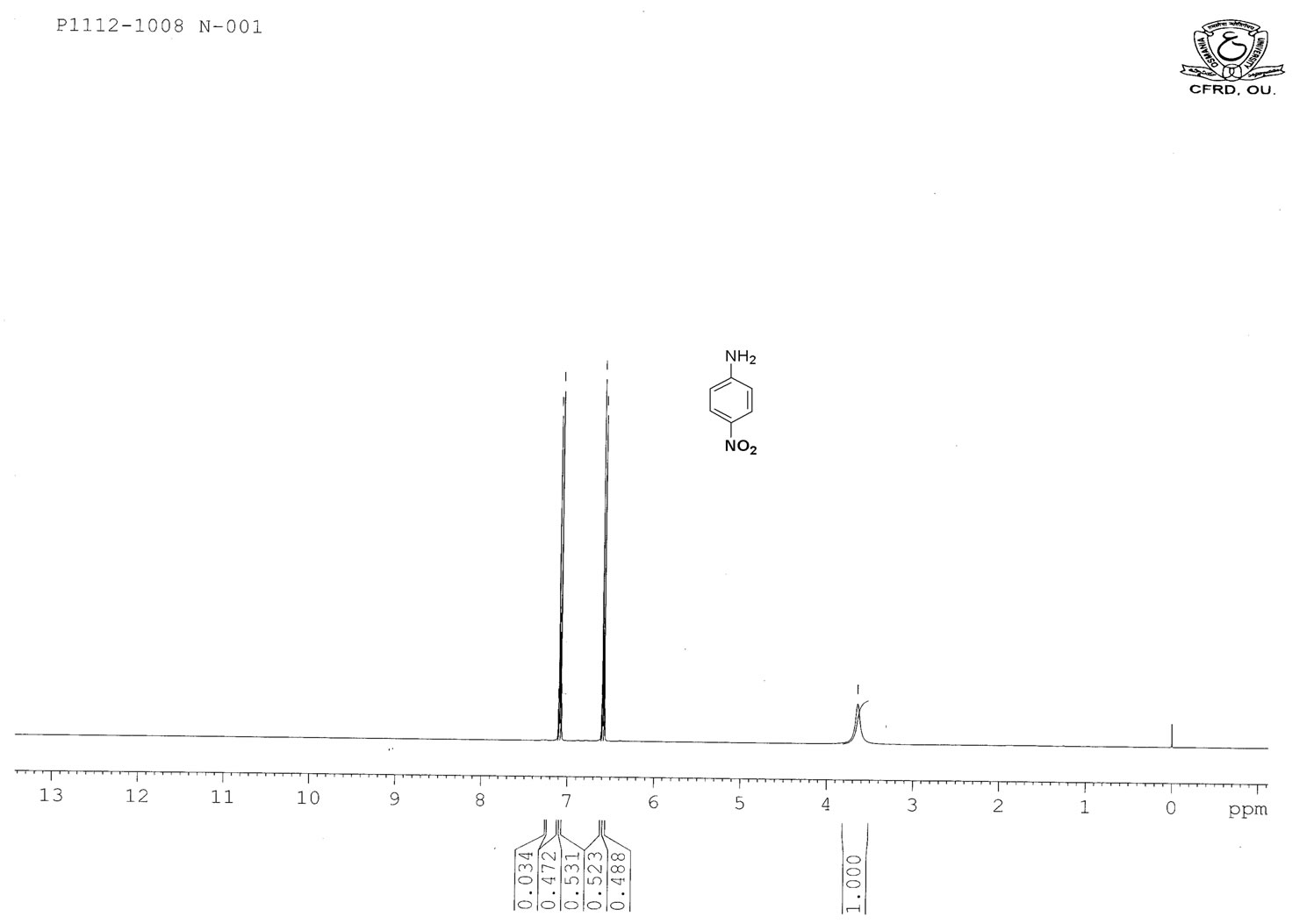
Figure S.6. HNMR Spectrum of 4-nitro aniline.
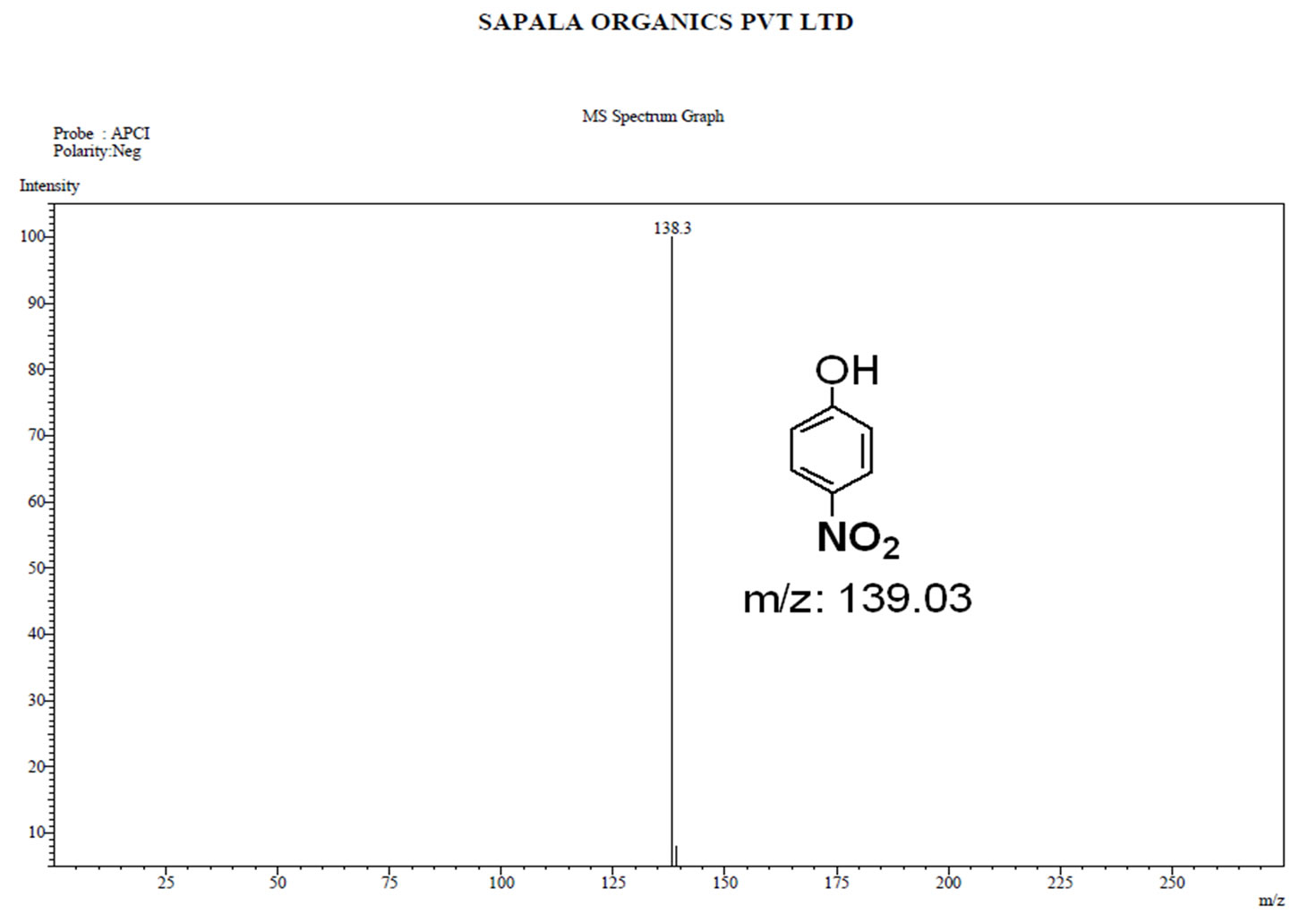
Figure S.7. Mass Spectrum of 4-nitro phenol.
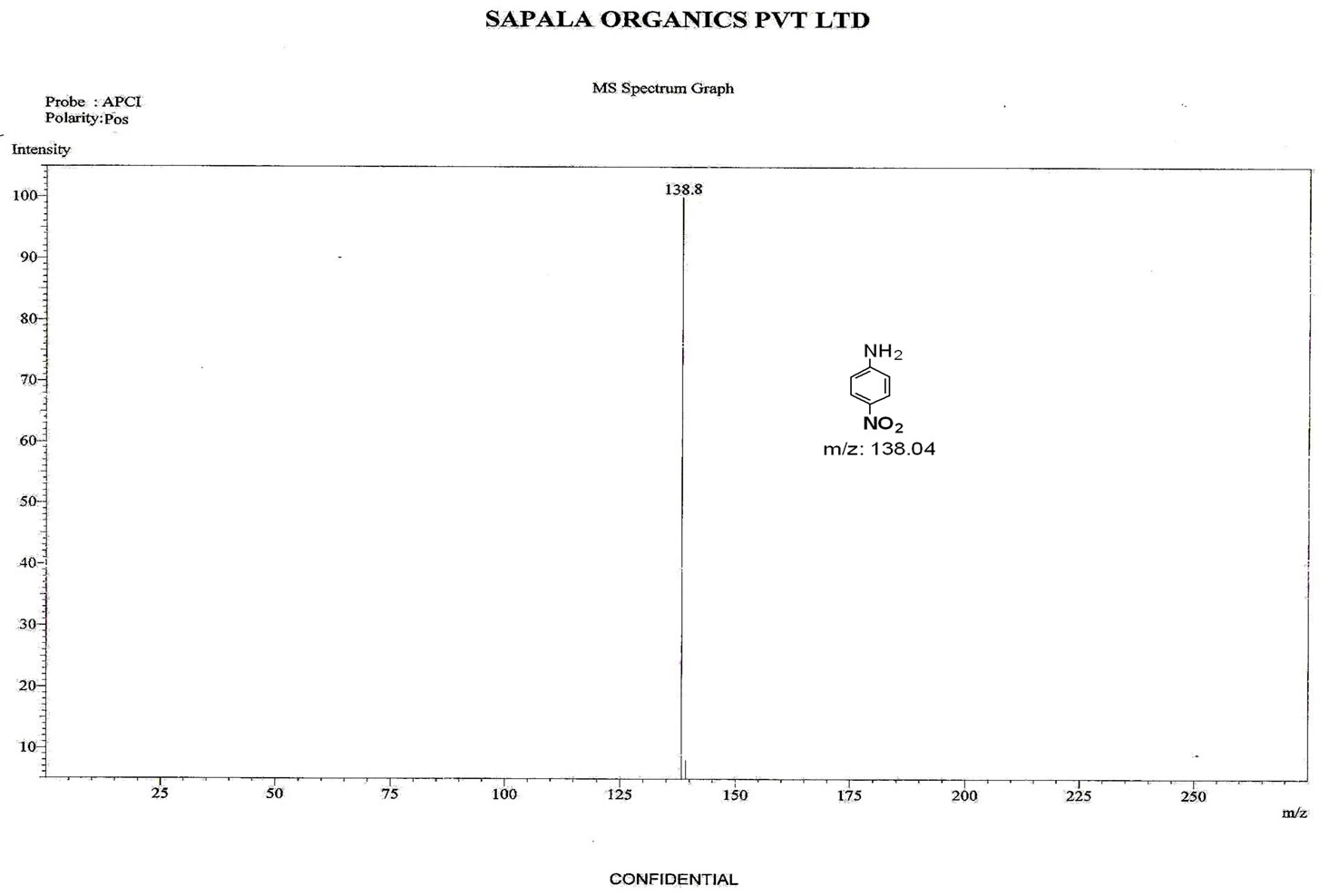
Figure S.8. Mass Spectrum of 4-nitro aniline.
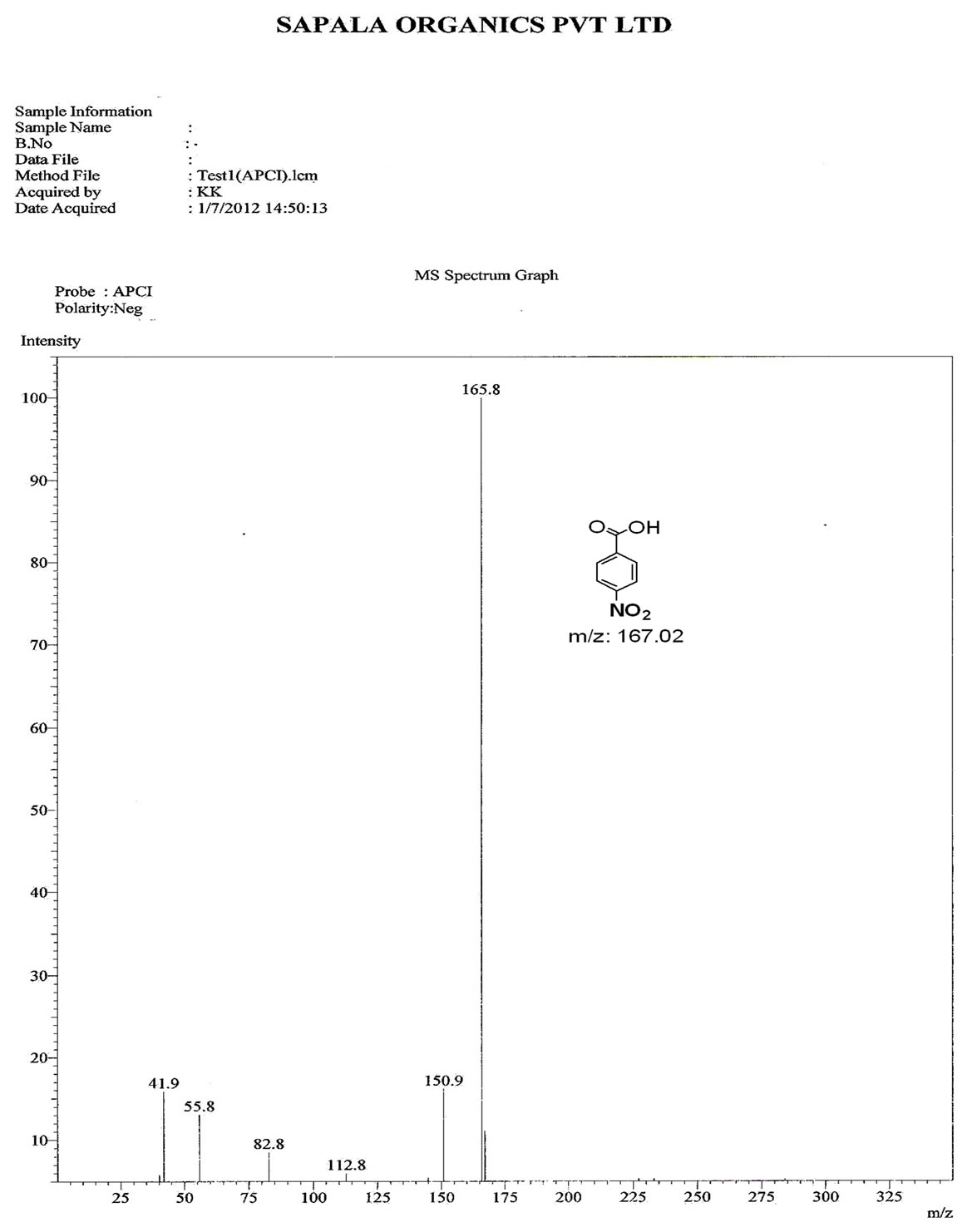
Figure S.9. Mass Spectrum of 4-nitro benzoic acid.
NOTES
*Corresponding author.