Journal of Analytical Sciences, Methods and Instrumentation
Vol.2 No.3(2012), Article ID:23286,4 pages DOI:10.4236/jasmi.2012.23026
Simultaneous Determination of Verapamil Hydrochloride and Gliclazide in Synthetic Binary Mixture and Combined Tablet Preparation by Chemometric-Assisted Spectroscopy
![]()
1Department of Pharmacy, Mahatma Jyoti Rao Phoole University, Jaipur, India; 2Department of Pharma Quality Assurance, Manipal College of Pharmaceutical Sciences Manipal University, Manipal, India.
Email: rahul.bhaskar03@gmail.com, *radhikabhaskar27@gmail.com, mahendraksagar@yahoo.com, vipinsaini31@rediffmail.com, km.bhat@manipal.edu
Received May 18th, 2012; revised June 14th, 2012; accepted June 25th, 2012
Keywords: Partial Least-Squares; Principle Components Regression; Spectroscopy; Verapamil Hydrochloride; Gliclazide
ABSTRACT
In this study, the simultaneous determination of verapamil hydrochloride and gliclazide in pharmaceuticals by chemometric approaches using UV spectrophotometry has been reported. Verapamil hydrochloride (VER) (Benzeneacetonitrile, α-[3-[[2-(3,4-dimethoxyphenyl) ethyl] methylamino]propyl]-3,4-dimethoxy-α-(1-methylethyl) hydrochloride) is an L-type calcium channel blocker of the phenylalkylamine class. It has been used in the treatment of hypertension, angina pectoris, and cardiac arrhythmia. Gliclazide (GLZ) (1-(Hexahydrocyclopenta[c]pyrrol-2(1H)-yl)-3-[(4-methylphenyl) sulphonyl]urea) is an oral hypoglycaemic (anti-diabetic) drug and is classified as a second generation sulfonylurea. Spectra of VER and GLZ were recorded at several concentrations within their linear ranges between wavelengths of 200 nm to 400 nm in 0.1N HCl. Partial least squares regression (PLS) and principle components regression (PCR) were used for chemometric analysis of data and the parameters of the chemometric procedures were optimized. The recoveries were satisfactory and statistically comparable. The method was successfully applied to pharmaceutical formulation, tablet, with no interference from excipients as indicated by the recovery study results. The proposed methods are simple, rapid and can be easily used in the quality control of drugs as alternative analysis tools.
1. Introduction
One of the basic problems in the dissolution testing of pharmaceutical preparations, containing two or more drugs is the simultaneous quantitative analysis without any chemical separation step. Earlier when chemometricing was not in frequent use, HPLC was one of the methods of choice of researchers for the analysis of the drugs showing an overlapped spectrum on u.v. spectrophotometry. But HPLC being an expensive and time consuming method, from the last few years, chemometric spectrophotometry is immensely attracting the analysts and playing an important role in the accurate detection of complex drug mixtures. In other words, chemometricing has added a new dimension in rapid and cost effective analysis of drugs having narrow difference in their lmax [1-3].
Several spectroscopic methods have been developed for the estimation of VER and GLZ individually and with other active pharmaceutical ingredients [4-9]. Nevertheless, to the best of our knowledge, no simultaneous estimation of these two compounds has been reported in combination using u.v. spectrophotometer.
Pharmaceutical processing and formulation often introduce various interferents (chemicals other than the drug under investigation) into the system. When performing quantification these interferents can disturb univariate analysis, but with multivariate analysis the quantification can still be performed. Several multivariate techniques of data analysis have been developed and used in the chemometric community by the researchers, out of which principal component regression (PCR) and partial least squares (PLS) regression are probably the most commonly applied ones. PLS regression is a supervised multivariate method with which quantitative analysis of multiple solid forms can be performed even if the differences between the spectra are minor [10]. The method involves a calibration step in which the relation between spectra and component concentrations is estimated from a set of reference samples, and a prediction step in which the results of the calibration are used to estimate the component concentrations in an unknown sample spectrum [11].
2. Experimental
2.1. Instrument, Reagents and Softwares
Elico SL 191 double beam UV-Visible Spectrophotometer, with 1 cm path length was used for the absorbance measurement. All the chemicals used were of analytical grade. Pure VER and GLZ were obtained from Kwality Pharmaceuticals, Amritsar.
The Matlab 7.5 with MVC1 toolbox and design expert 8.0.4 softwares were used for the statistical treatment of the data and application of various multivariate methods.
2.2. Procedure
2.2.1. Preparation of Standards
1 mg/ml VER and GLZ stock solutions were prepared by dissolving accurately weighed amounts of finely powdered pure VER and GLZ in small quantity of methanol and the final volume was made up with 0.1N HCl for the respective samples. Suitably diluted samples from each stock were utilized for lmax determination of individual component followed by serial dilution of stocks with 0.1N HCl to obtain the aliquots falling in linearity calibration range.
2.2.2. Standard Solutions for Multivariate Calibration
The calibration and test mixtures were prepared by mixing the VER and GLZ solutions in different ratios varying within their individual linearity range viz. 0 - 60 µg/ml and 0 - 18 µg/ml respectively. The concentration of combinations was decided by design expert 8.0.4 software under general factorial design. A total of 26 sets were prepared out of which 14 were calibration sets (Table 1) whereas, the rest 12 served as validation sets (Table 2). All the mixtures were scanned at 220 - 300 nm range. The absorbance below 220 nm and above 300 nm was not taken under consideration due to too much of noise and diminished responses respectively.
2.2.3. Sample Preparation
Commercial tablets of VER and GLZ were analyzed. The tablets were processed as follows: at least 10 tablets were taken for each and finely crushed to powder in two separate mortar and pestle. A suitable amount of the obtained powders of each drug in possible highest and lowest concentration ratios were separately weighed, dissolved in methanol, sonicated for 20 minutes, and filtered through a 0.5 µm membrane filter. Each sample solution was prepared in triplicate and measured in random order.
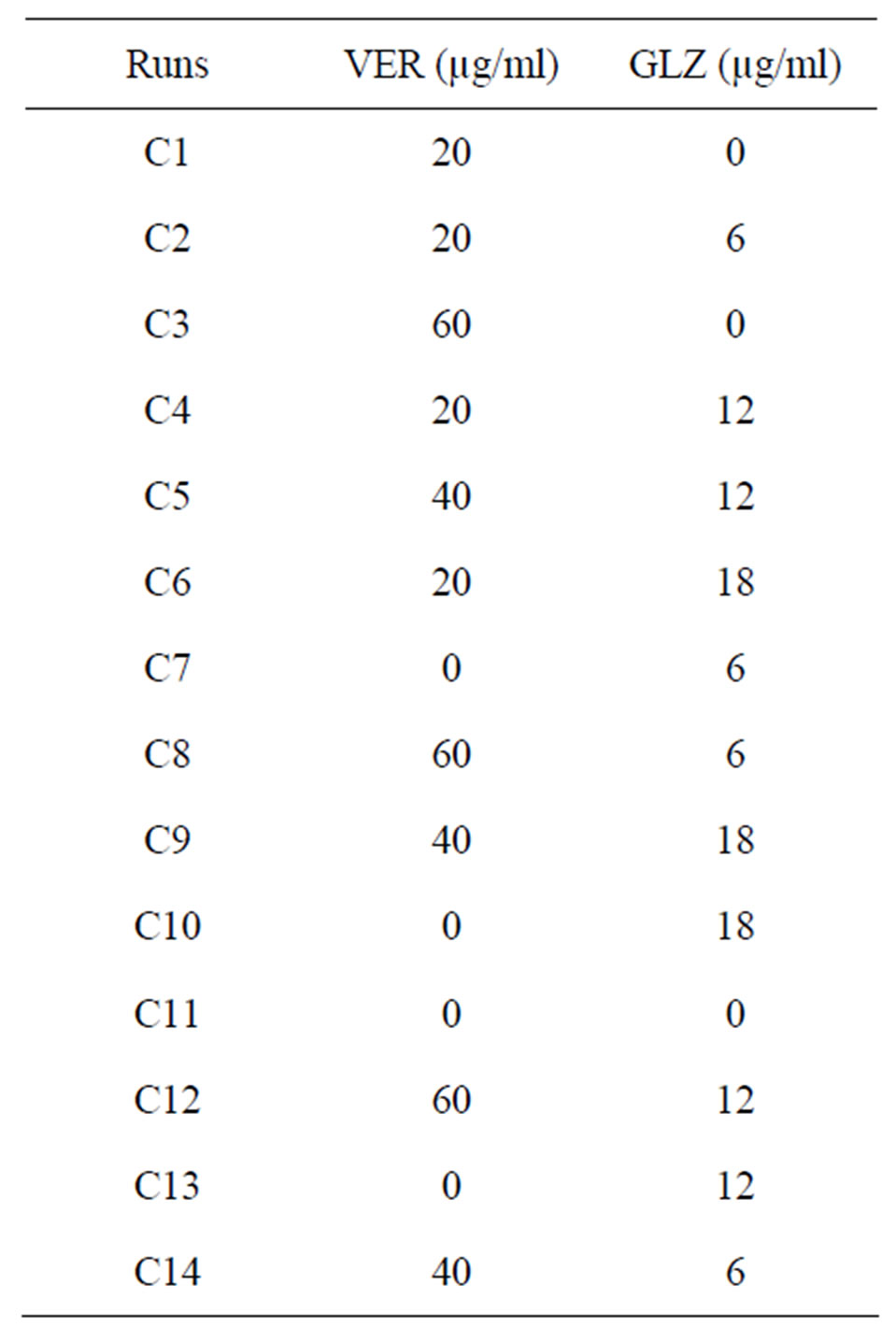
Table 1. Calibration set composition.
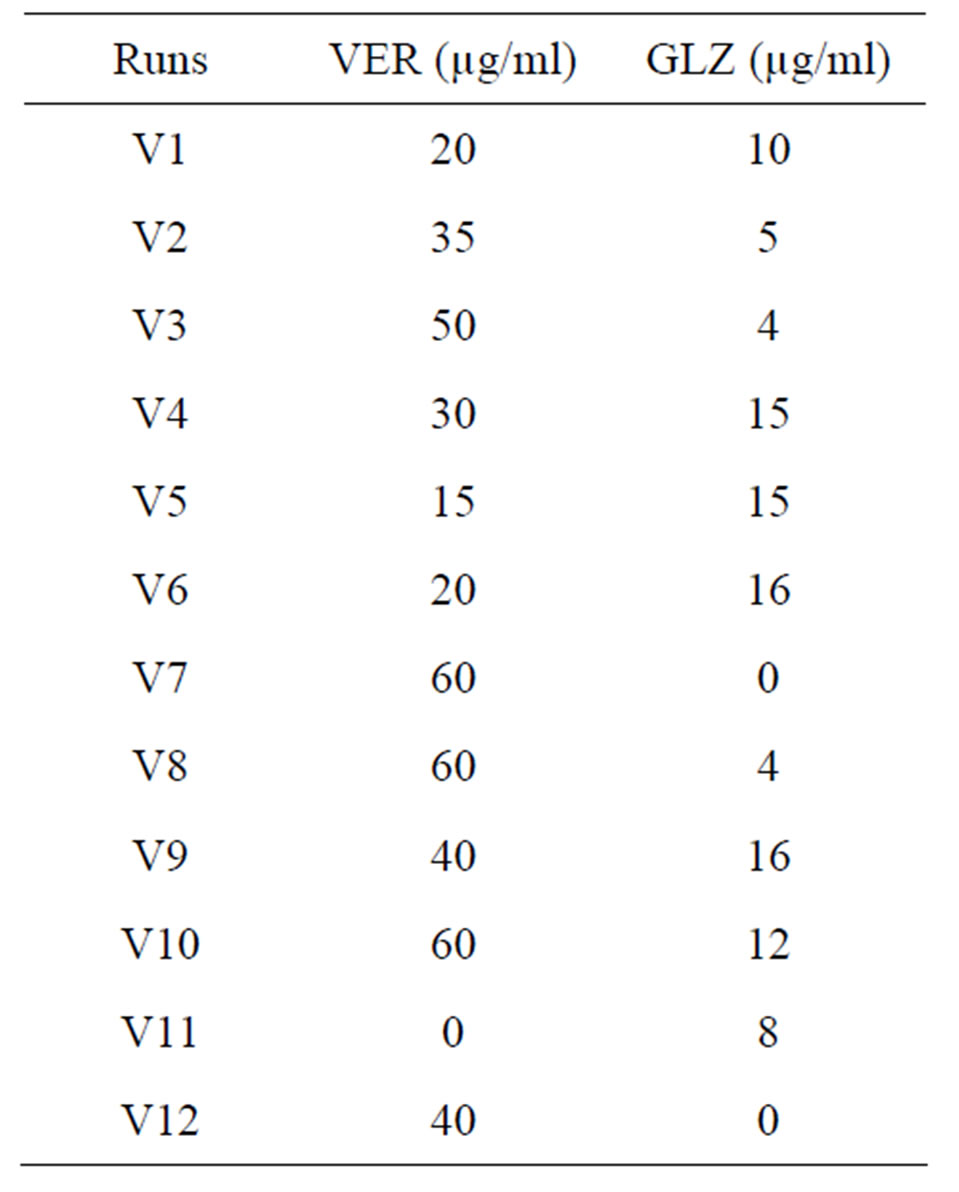
Table 2. Validation set composition.
3. Theory
PCR method comprises of several steps, the first one is to identify a data matrix which can be denoted by A. The matrix A represents a set of absorbance for samples as row × column matrix. Next step is to calculate the singular value decomposition (SVD) for matrix A, which result into U matrix (left singular value), S matrix (singular value matrix) and the V matrix (right singular value) (Equation (1))
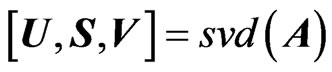 (1)
(1)
Third step involves generation of score matrix and loading matrix. Loading matrix is nothing but the right singular matrix or V matrix whereas, score matrix (T) can be calculated as follows (Equation (2)):
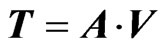 (2)
(2)
Data matrix A is estimated by the equation
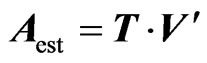 (3)
(3)
In fourth step we can determine the regression vector by using decomposed matrices along with matrix c (concentration of the samples used in generation of matrix A). Last step is to utilize the regression vector for determination of unknown concentration with the help of Equation (4) against test absorbance matrix.
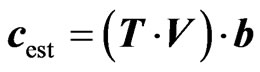 (4)
(4)
where, c is the concentration vector and b is the regression vector.
PLS also follows the similar basic steps as that of PCR except that it uses concentration information during the decomposition process. This causes spectra containing higher constituent concentrations to be weighted more heavily than those with low concentration resulting as high concentration information as possible into the first few loading vectors [12].
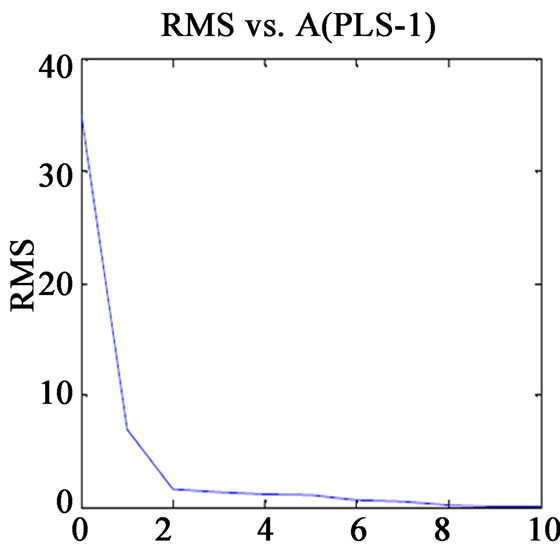
Before finalising the calibration data, to avoid over fitting, the optimum number of latent variables (Figure 1) should be selected by applying the cross validation method, leaving one sample at a time [13]. The usual statistical parameters giving an indication of the quality of fit of all data are the root mean square difference (RMSECV), square of the correlation coefficient (R2) and relative error of prediction (REP%). The expressions of these parameters are:
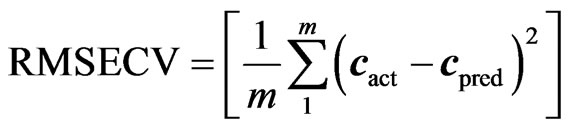 (5)
(5)
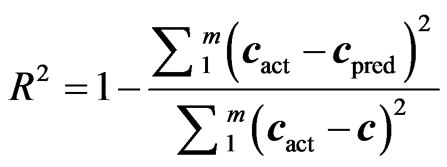 (6)
(6)
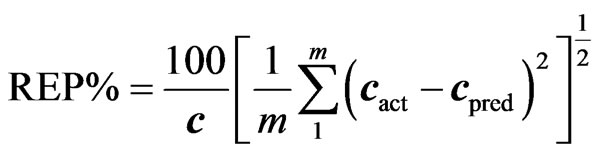 (7)
(7)

where cact and cpred are the actual and predicted concentrations during the cross validation process, m is number of samples used in cross validation and validation [11].
 (a)
(a)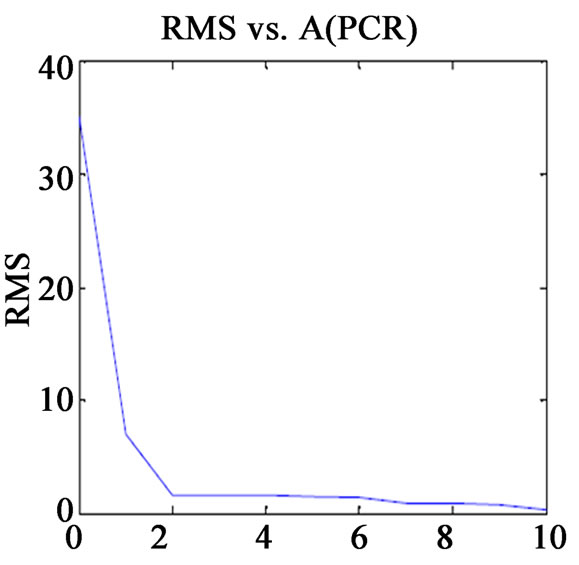 (b)
(b)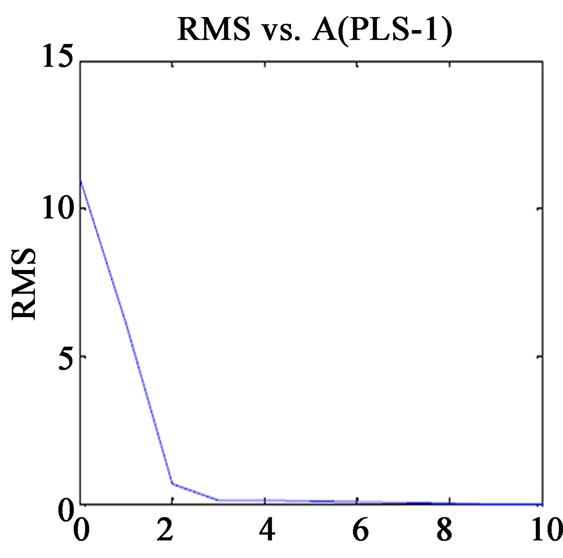 (c)
(c)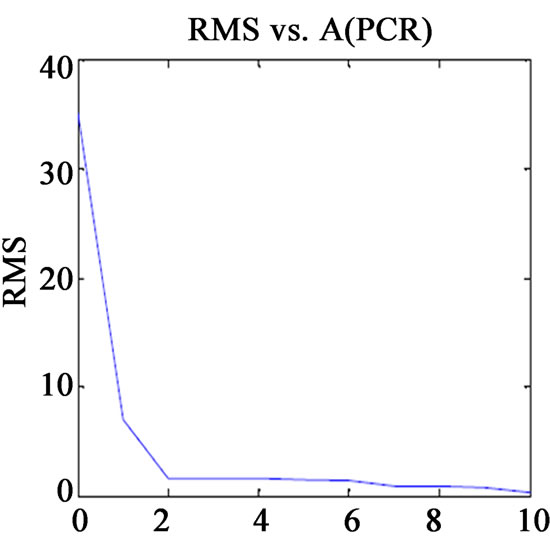 (d)
(d)
Figure 1. Plot of RMS(CV) vs. factor number for calibration set prediction using cross validation of (a) VER PLS-1; (b) VER PCR; (c) GLZ PLS-1; (d) GLZ PCR.
The goodness of data fit can be visualized in Figure 2.
Along with the above said statistical formulae, another preferred method for assessing the relative accuracy of the studied models is the linear regression analysis of actual verses predicted data by comparing the results of the estimated slope and intercept with their ideal value of 1 and 0. If the point (1, 0) is inside the EJCR (elliptical joint confidence region) for cross validation data, it can be concluded that constant and proportional bias are absent (Figure 3).
The sensitivity for a given analyte such as k can be calculated as
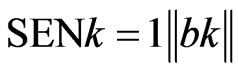
where  indicates the Euclidian norm and bk is the vector of regression coefficients. Better insight is furnished by the analytical sensitivity, defined by
indicates the Euclidian norm and bk is the vector of regression coefficients. Better insight is furnished by the analytical sensitivity, defined by

where 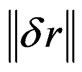 is a measure of the instrumental noise. It helps in the comparison of analytical methods regardless of the specific technique, equipment and scale employed. Moreover, it establishes the minimum concentration difference (γ−1k), which is statistically discernible by the method along the dynamic range.
is a measure of the instrumental noise. It helps in the comparison of analytical methods regardless of the specific technique, equipment and scale employed. Moreover, it establishes the minimum concentration difference (γ−1k), which is statistically discernible by the method along the dynamic range.
Finally, the limit of detection can be calculated as [14]
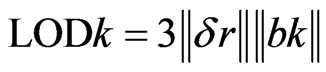
4. Result and Discussion
4.1. UV-Vis Spectra of VER, GLZ and Mixture
Figure 4 shows the individual absorption spectra of VER and GLZ along with their mixture in 0.1N HCl between 200 and 300 nm. Though the lmax of GLZ and VER were found to be 226 nm and 278 nm resp. But since VER drugs show a strong overlap at the lmax of GLZ therefore, conventional univariate spectrophotometric methods cannot be applied for resolving this mixture.
4.2. PLS-1 and PCR Results
The cross validation and validation statistical parameters obtained after applying PLS and PCR to the spectrophotometric data are shown in Table 3, showing reasonably low absolute and relative root-mean square errors (RMSECV and REP%, respectively). The set of 12 validation samples prepared as described above was analysed by the proposed procedure. The results suggest that the present method is accurate in concern to the validation samples, as suggested by the low RMSE and REP value for this validation set.
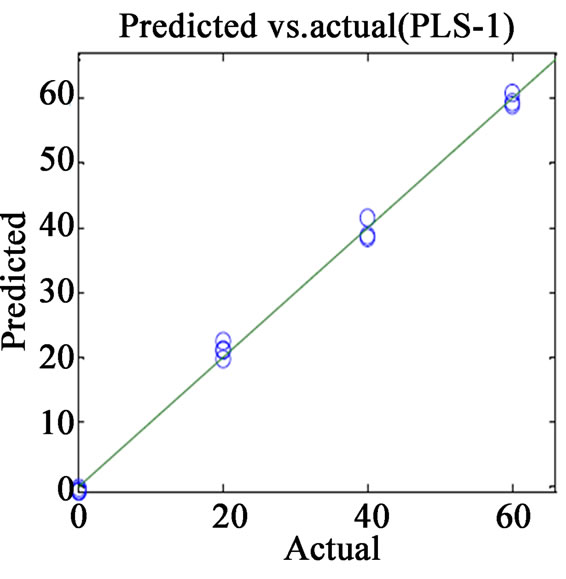 (a)
(a)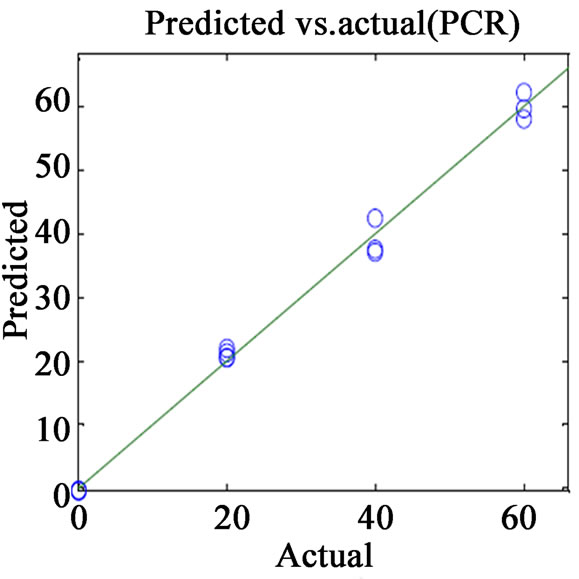 (b)
(b) (c)
(c)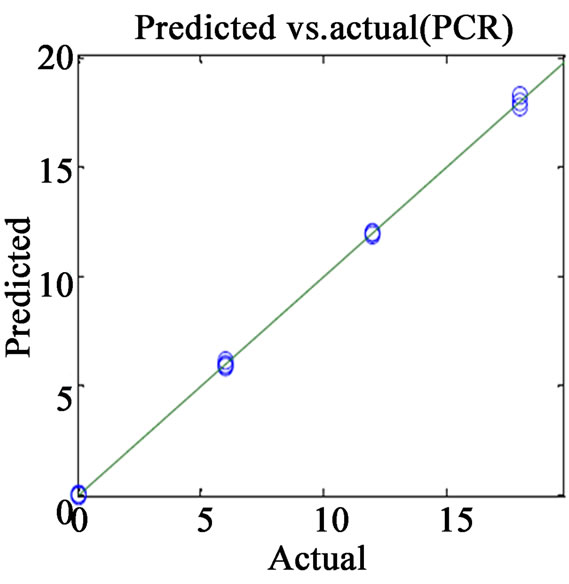 (d)
(d)
Figure 2. Plots of actual vs. predicted values for (a) VER PLS-1; (b) VER PCR; (c) GLZ PLS-1; (d) GLZ PCR.
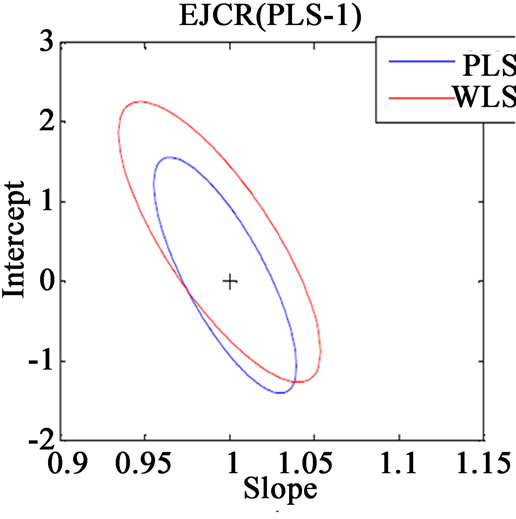 (a)
(a)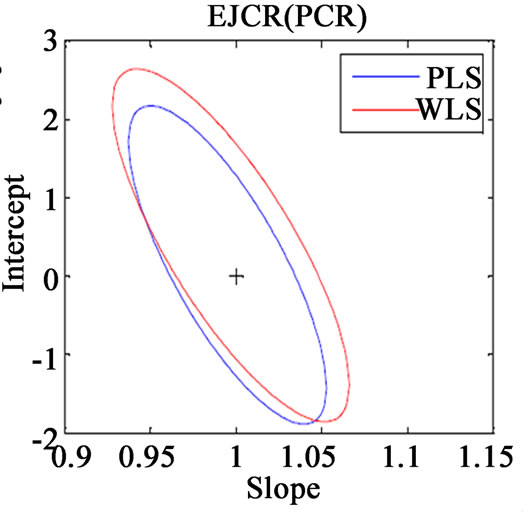 (b)
(b)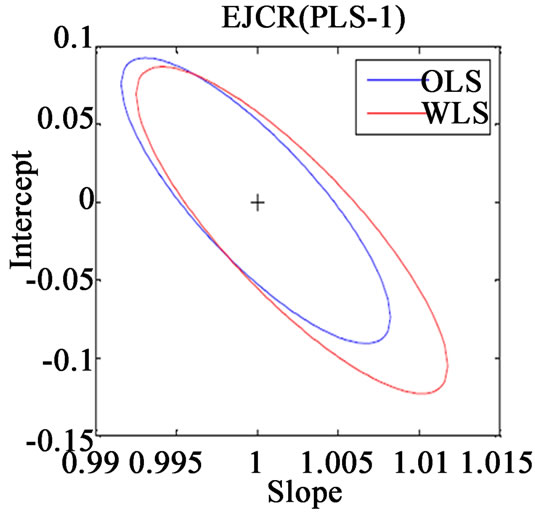 (c)
(c)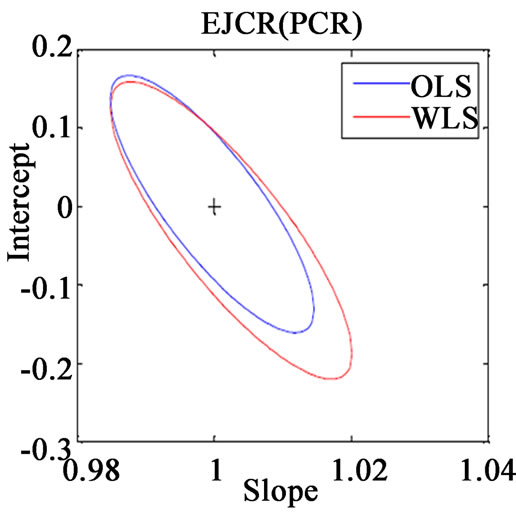 (d)
(d)
Figure 3. Ellipticle joint confident region for slope and intercept corresponding to regressions of the actual vs. predicted concentrations of (a) VER PLS-1, (b) VER PCR, (c) GLZ PLS-1, (d) GLZ PCR.
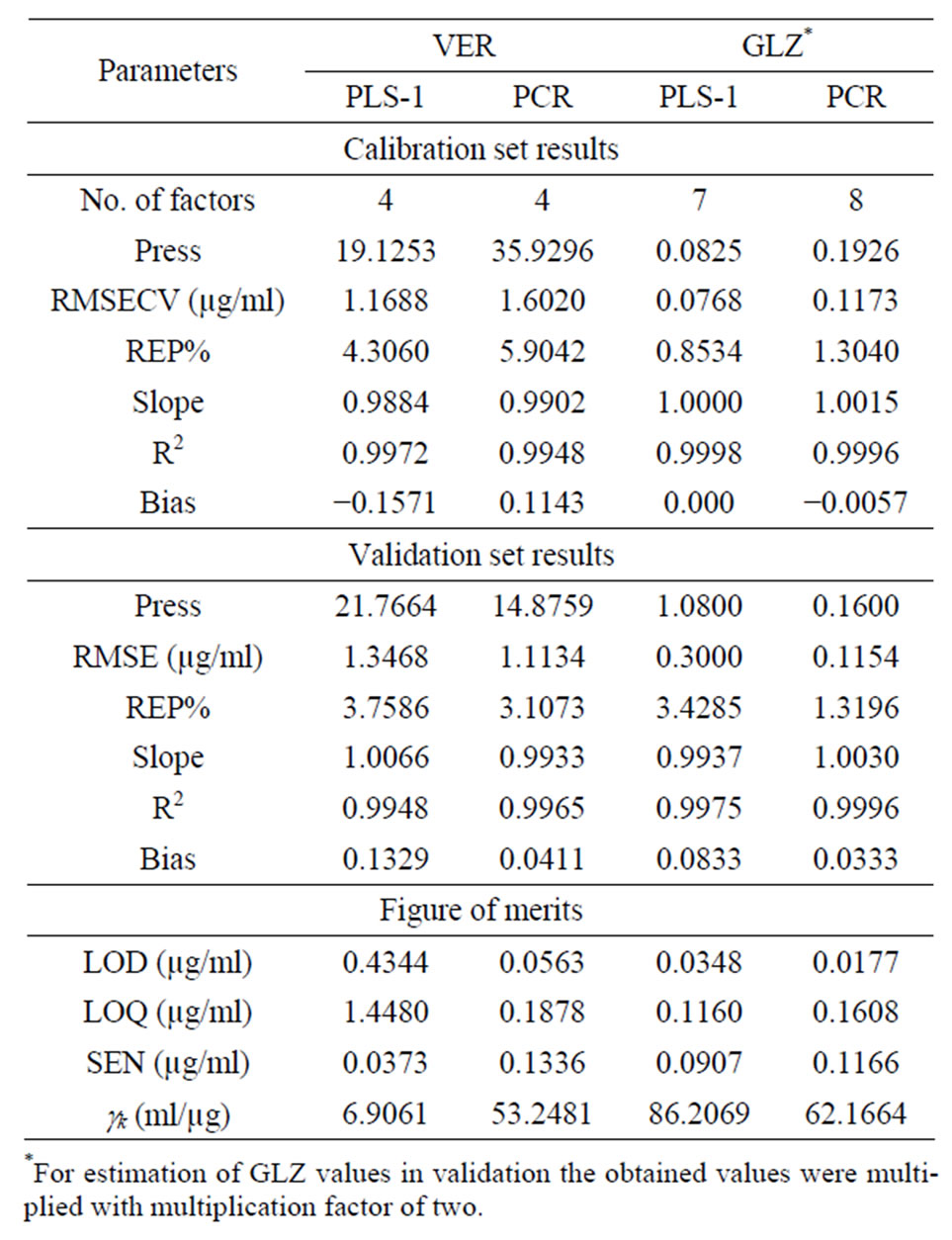
Table 3. Statistical parameters for the optimised models.
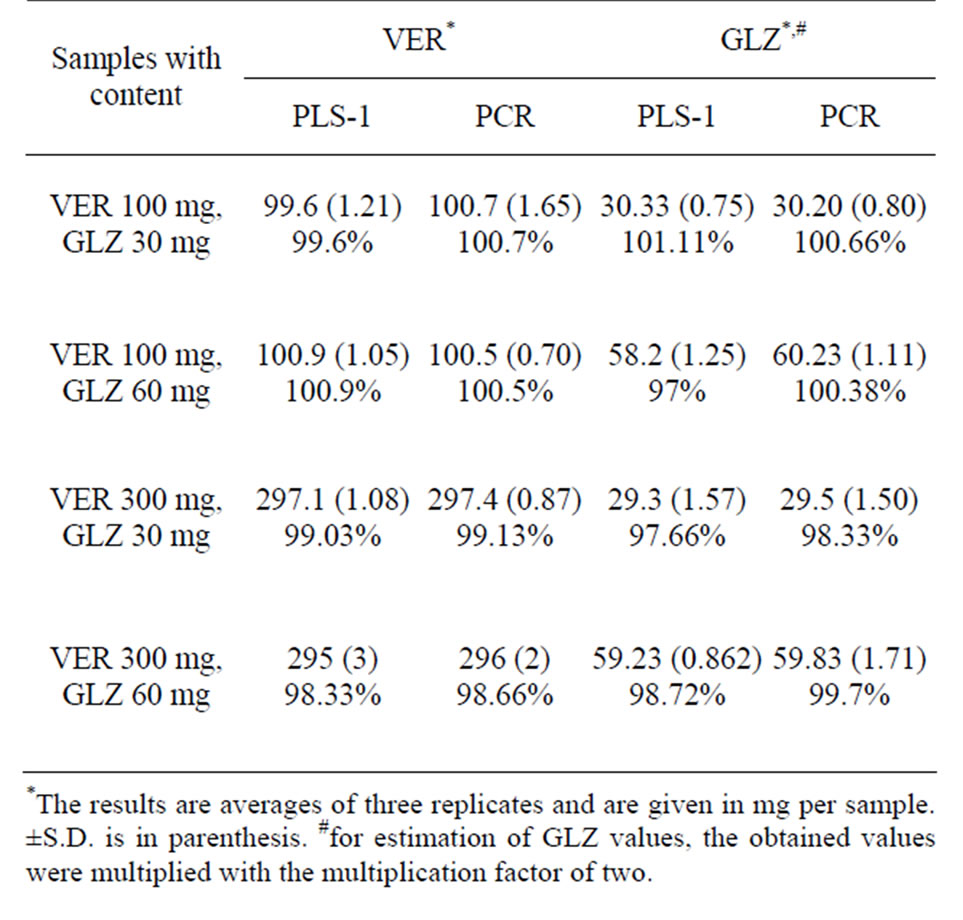
Table 4. Prediction results on recovery samples.
4.3. Analysis of Commercial Sample
Commercial mixture products were analysed using the proposed spectrophotometric methods. Results are summarised in Table 4. As can be seen, satisfactory results were obtained by the proposed methods.
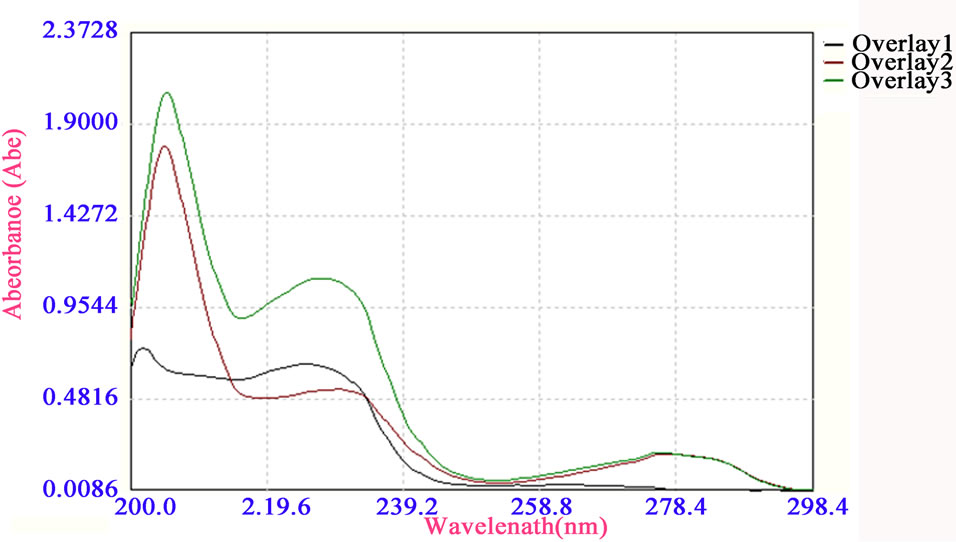
Figure 4. Overlay of VER, GLZ and Mixture.
5. Conclusion
A comparative study of the use of PLS-1 and PCR for the separation and simultaneous estimation of VER and GLZ in a binary mixture has been accomplished, showing that this method provide a clear example of the high resolving power of these techniques. In other words, almost comparable results were obtained for these two drugs in both synthetic and commercial mixture applications by PLS and PCR. The results obtained confirm the suitability of the proposed method for accurate analysis of verapamil hydrochloride and gliclazide in pharmaceutical preparations. These methods were applied directly to the commercial mixture preparations without previous treatment. In addition the proposed methods are suitable for application without interference of the excipients.
6. Acknowledgements
Our sincere thanks to B. R. Nahata College of Pharmacy, Mandsaur for providing the laboratory facilities.
REFERENCES
- E. Dinte, I. Tomuta, E. M. Mut, R. I. Iovanov and S. Leucuta, “Chemometric Methods for the Simultaneous Assay of Chloramphenicol, Chlorhexidine and Metronidazole during in Vitro Dissolution of Drugs from Mucoadhesive Buccal Gels,” Farmacia, Vol. 58, 2010, pp. 572-582.
- N. Benoudjit, F. Melgani and H. Bouzgou, “Multiple Regression Systems for Spectrophotometric Data Analysis,” Chemometrics and Intelligent Laboratory Systems, Vol. 95, No. 2, 2009, pp. 144-149. doi:10.1016/j.chemolab.2008.10.001
- R. Sawant, R. Bhangal, R. Joshi and P. Lanke, “Validated Spectrophotometric Methods for Simultaneous Estimation of Paracetamol, Domperidone and Tramadol HCl in Pure and Tablet Dosage Form,” Journal of Chemical Metrology, Vol. 4, 2010, pp. 21-27.
- M. S. Araynel, N. Sultana and A. Z. Mirza, “Simultaneous Determination of Gliquidone, Pioglitazone Hydrochloride, and Verapamil in Formulation and Human Serum by RP-HPLC,” Journal of Chromatographic Science, Vol. 49, No. 2, 2011, pp. 114-117. doi:10.1093/chrsci/49.2.114
- L. Chytil, B. Strauch, J. Cvacka, V. Maresova and O. Slanar, “Determination of Doxazosin and Verapamil in Human Serum by Fast LC-MS/MS: Application to Document Non-Compliance of Patients,” Journal of Chromatography B, Vol. 878, No. 30, 2010, pp. 3167-3173. doi:10.1016/j.jchromb.2010.09.032
- P. N. Dhabale and C. R. Seervi, “Simultaneous UV Spectrophotometric Method for Estimation of Gliclazide and Metformine Hydrochloride in Tablet Dosage Form,” International Journal of Chemical Technology Research, Vol. 2, 2010, pp. 813-817.
- G. Rathinavel, U. U. Nath, J. Valarmathy, L. S. Joshua, C. S.Thanuja, M. Ganesh, T. S. Kumar and R. Priyadarsini, “RP-HPLC Method for the Simultaneous Estimation of Rosiglitazone and Gliclazide in Tablets,” E-Journal of Chemistry, Vol. 6, 2009, pp. 1188-1192.
- Y. K. Aggarwal, P. J. Gogoi, K. Manna, H. G. Bhatt and V. K. Jain, “A Supercritical Fluid Chromatography/Tandem Mass Spectrometry Method for the Simultaneous Quantification of Metformin and Gliclazide in Human Plasma,” Journal of Pharmaceutical Science, Vol. 72, 2010, pp. 50-57.
- G. Venkatesh, V. Ramanathan, S. M. Mansor, N. K. Nair, M. A. Sattar, S. L. Croft and V. Navaratnam, “Development and Validation of RP-HPLC-UV Method or Simultaneous Determination of Buparvaquone, Atenolol, Propranolol, Quinidine and Verapamil: A Tool for the Standardization of Rat in Situ Intestinal Permeability Studies,” Journal of Pharmaceutical Biomedical Analysis, Vol. 43, No. 4, 2007, pp. 1546-1551. doi:10.1016/j.jpba.2006.11.013
- Y. Ni, Y. Wang and S. Kokot, “Multicomponent Kinetic Spectrophotometric Determination of Pefloxacin and Norfloxacin in Pharmaceutical Preparations and Human Plasma Samples with the Aid of Chemometrics,” Spectrochimica Acta Part A, Vol. 70, No. 5, 2008, pp. 1049-1059. doi:10.1016/j.saa.2007.10.013
- M. J. Culzoni, M. D. Zan, J. C. Robles, V. E. Mantovani and H. C. Goicoechea, “Chemometrics-Assisted UVSpectroscopic Strategies for the Determination of Theophylline in Syrups,” Journal of Pharmaceutical Biomedical Analysis, Vol. 39, No. 5, 2005, pp. 1068-1074. doi:10.1016/j.jpba.2005.06.009
- H. Mark and J. Workman, “Chemometrics in Spectroscopy,” Academic Press, London, 2007.
- D. M. Haaland and E. V. Thomas, “Partial Least-Squares Methods for Spectral Analyses,” Analytical Chemiatry, Vol. 60, No. 11, 1988, pp. 1193-1202. doi:10.1021/ac00162a020
NOTES
*Corresponding author.