Open Access Library Journal
Vol.03 No.04(2016), Article ID:69192,12 pages
10.4236/oalib.1102398
Analytical Procedures and Method Validation for Determination of Chlorimuron Ethyl in Sediments
Ramom Rachide Nunes1*, Bruno Henrique Froes Santos2, Maria Olímpia de Oliveira Rezende1
1University of São Paulo, São Carlos Institute of Chemistry, São Carlos, Brazil
2School of Engineering of São Carlos, São Carlos School of Engineering, São Carlos, Brazil

Copyright © 2016 by authors and OALib.
This work is licensed under the Creative Commons Attribution International License (CC BY).
http://creativecommons.org/licenses/by/4.0/


Received 4 April 2016; accepted 18 April 2016; published 21 April 2016

ABSTRACT
The present work describes the validation of an analytical method for chromatographic determination of chlorimuron ethyl herbicide (compound of sulphonylurea group) in sediments, in according with Brazilian legislation, following the guidance and normative documents of INMETRO (DOQ-CGCRE-008) and ANVISA (Resolution RE no. 899-2003). The results show that the chromatographic method demonstrated to be selective, accurate, precision and linear over the concentration range of 5.0 - 1000.0 μg∙L−1. Besides, limits of quantitation and determination were satisfactory, 29.48 and 8.84 μg∙L−1, both determined by calibration curve. The method proposed also showed good results for assays of recovery, with confidence intervals between 80 and 100%, approximately, with accuracy and precision.
Keywords:
Chlorimuron Ethyl, Sulphonylurea, Validation of Analytical Method, HPLC-UV
Subject Areas: Analytical Chemistry, Environmental Chemistry, Environmental Sciences

1. Introduction
O aumento crescente na demanda por alimentos e insumos agrícolas tem impulsionado a utilização cada vez maior de pesticidas. Plantas, insetos, bactérias e outros organismos são uma parte natural de um ecossistema. No ambiente, esses organismos podem ser benéficos de muitas maneiras mas, também, podem se tornar uma praga. Nas práticas agrícolas as ervas daninhas, pulgões, larvas, entre outros, são exemplos de interferentes naturais que precisam ser controlados. Este controle geralmente se dá pelo uso de pesticidas [1] [2] .
Dentre as classes de pesticidas, destacam-se os herbicidas. Atualmente, 12.2% do mercado de herbicidas pertence ao grupo químico das sulfonilureias, que ocupa o segundo lugar no setor [3] .
A estrutura geral das sulfonilureias baseia-se na fórmula R1-SO2-NH-CO-NH-R2, em que o substituinte R1 pode ser uma cadeia alifática, aromática ou um grupo heterocíclico conectado por uma ponte sulfonilureia ao substituinte R2, que pode ser uma pirimidina ou triazina heterocíclica [4] .
Neste trabalho, a sulfonilureiaestudada foi o o etilclorimuron (Figure 1), herbicida largamente utilizado no controle de ervas daninhas em plantações de soja. No Brasil, o herbicida etilclorimuron foi licenciado pelo governo federal há 27 anos, conforme registrado no Diário Oficial da União, passando a ser de uso permitido em atividades agropecuárias [5] .
Na análise de impactos ambientais os sedimentos são de relevante importância, pois atuam como depósitos de muitas substâncias químicas, especialmente os elementos tóxicos e compostos orgânicos, tais como os pesticidas. Sedimentos são camadas de partículas minerais e orgânicas, frequentemente de granulometria fina, encontradas em contato com os corpos de águas naturais como lagos, rios e oceanos [1] .
Nas últimas décadas, diversas técnicas analíticas têm sido utilizadas no isolamento e quantificação de pesticidas. Dentre estas, a cromatografia vem sendo empregada na determinação de herbicidas e seus metabólitos em matrizes ambientais: solo, água, sedimentos etc. [6] .
Separações difíceis são, geralmente, alcançadas mais facilmente pela cromatografia líquida de alta eficiência (HPLC). Mesmo apresentando limitações como alto custo de instrumentação e operação, a cromatografia líquida tem sido cada vez mais usada em análises de resíduos de pesticidas devido a sua versatilidade. Pode ser aplicada para compostos orgânicos e inorgânicos, iônicos ou covalentes [7] .
Quando se trabalha com cromatografia e se pensa em verificar se um determinado resultado é confiável ao analito, todo sistema ao redor dessemétodo deve ter, também, algum sentido de confiabilidade. Ter um método enquadrado em um nível de validação significa agregar confiança e credibilidade aos dados obtidos [8] [9] .
Validar um método analítico é dar ao mesmocredibilidade e confiança. No caso de um sistema de análise de substâncias químicas, a validação objetiva, principalmente, assegurar que o sistema funcione adequadamente dentro das condições de análise para as quais foi validado [9] .
O processo de validação pode ser regido por documentos orientativos e/ou normativos publicado por órgãos e autarquias governamentais. Neste trabalho, os critérios de validação serão discutidos em conformidade a dois documentos ligados a órgãos federais do governo brasileiro: o INMETRO (Instituto Nacional de Metrologia, Qualidade e Tecnologia) [10] e a ANVISA (Agência Nacional de Vigilância Sanitária) [11] .
Assim, o presente trabalho teve como objetivo validar um método analítico para determinação cromatográfica do herbicida etilclorimuron, composto da classe das sulfonilureias, em sedimentos. Foram avaliados os seguintes parâmetros: seletividade, linearidade, faixas de trabalho e lineares, limites de detecção e quantificação, sensibilidade, recuperação, precisão e robustez.
2. Parte Experimental
2.1. Materiais
Para o preparo das soluções foram utilizadas vidrarias e acessórios volumétricos Classe A da marca Carvalhaes (Porto Alegre, Brasil) calibrados conforme critérios da norma ISO 17.025 pela empresa ELUS Instrumentação (São Paulo, Brasil), que é credenciada pelo INMETROe acreditada pela RBC (Rede Brasileira de Calibração) [12] .

Figure 1. Estrutura química do herbicida etilclorimuron.
O padrão analítico do herbicida etilclorimuron (CAS 90982-32-4), 97% m/m (Sigma-Aldrich, Alemanha), foi determinado e quantificado via cromatografia líquida de alta eficiência com detector de ultravioleta (HPLC-UV). Nas determinações cromatográficas utilizou-se um equipamento Shimadzu (Kioto, Japão), modelo SCL-10A com: detector UV-Vis SPD-20A, degaseificador DGU-20A5, bomba LC-20AT e sistema de controle CBM-20A.
Para determinação da seletividade, o espectro de UV-Vis do herbicida etilclorimuron foi obtido em um espectrofotômetro Jasco V630 (Oklahoma, Estados Unidos).
2.2. Amostragem Dos Sedimentos
As amostras de sedimento analisadas foram coletadas em corpos d’água lênticos distribuídos em três ambientes distintos ao longo de uma paisagem agrícola. A amostragem foi realizada em poças, brejos, lagoas, dutos e canaletas e reservatórios, entre outros. A região analisada pertence ao entorno da cidade de Luís Antônio, São Paulo (SP), Brasil.As coletas foram realizadas de acordo com o Guia Nacional de Coleta e Preservação de Amostras da Agência Nacional de Águas (ANA) e Companhia de Tecnologia de Saneamento Ambiental(CETESB) [14] .
As amostras foram nomeadas em função do uso dado ao solo (Figure 2):

Figure 2. Município de Luís Antônio (SP), indicando as regiões onde foram realizadas as amostragens dos sedimentos: zona agrícola (ZA), zona pecuária (ZP) e estação ecológica (EE).
i. Zona Agrícola (ZA): ambiente de cultivo extensivo e latifundiário de cana-de-açúcar além de outras culturas rotativas de menor importância econômica (47 pontos de coleta);
ii. Zona Pecuária (ZP): ambiente de pastagem para o gado de corte e leiteiro. Região localizada na confluência com o perímetro urbano adjacent (20 pontos de coleta);
iii. Estação Ecológica (EE): ambiente de referência com reduzida ação antrópica, coberto por cerradão, mata estacional semi-decídua, localizado na Estação Ecológica e Experimental de Jataí (17 pontos de coleta).
As escolha destes diferentes tipos de sedimentos deveu-se às propriedades e características distintas dessas matrizes analítico-ambientais. A fim de verificar a influência das características físicas e químicas dos sedimentos na validação do método, determinaram-se as propriedades: umidade, capacidade de troca catiônica (CTC), classe textural, teores de matéria orgânica (MO) e de ácidos húmicos (AH). Para isso seguiram-se as metodologias sugeridas pela EMBRAPA (Empresa Brasileira de Pesquisa Agropecuária) e pela Sociedade Internacional de Substâncias Húmicas (IHSS) [15] .
2.3. Validação Do Método Analítico
2.3.1. Parâmetros Cromatográficos
Os parâmetros cromatográficos em validação foram selecionados após testes preliminares. São eles:
i. Coluna: RP-18 5 um de 250 × 4.6 mm (Agilent Zorbax, Estados Unidos);
ii. Composição da fase móvel: 60% de CH3CN + 40% de solução H3PO4 0.1%;
iii. Eluição isocrática;
iv. Fluxo da fase móvel: 1.4 mL∙min−1;
v. pHSolução da Fase Móvel/H3PO4: 2.0;
vi. pHFase Móvel/CH3CN + H3PO4: 2.7;
v. Volume de injeção: 20.0 μL (volume da alça do injetor);
vi. Comprimento de onda do detector UV: 235 nm;
vii. Tempo de análise: 4 min.
2.3.2. Preparo Da Solução Estoque E Soluções Iniciais
A partir do padrão analítico do herbicida etilclorimuron (sólido) preparou-se um volume de 100.0 mL de uma solução estoque com concentração de 100.0 mg∙L−1 em acetonitrila (Tedia, Brasil).
A partir desta solução estoque foram preparadas duas soluções iniciais com volume de 10.0 mL e concentrações de 10.0 mg∙L−1 (solução Si) e 1.0 mg∙L−1 (solução Sii).
2.3.3. Seletividade
Os ensaios de seletividade foram avaliados por meio da comparação dos resultados obtidos em amostras isentas da substância de interesse (etilclorimuron) e dos sedimentos adicionados do analito.
Os ensaios para análise da seletividade foram realizados em duas etapas. Na primeira, o herbicida etilclorimuron (solução inicial Sii) foi determinado por HPLC-UV seguindo os parâmetros cromatográficos em validação. Na segunda etapa, repetiu-se o mesmo procedimento na análise dos sedimentos extraídos em acetonitrila.
Para obtenção do extrato dos sedimentos, adicionou-se 1 g de sedimento em 10 mL de acetonitrila. O conteúdo foi mantido sob agitação orbital durante 30 minutos e, em seguida, a matriz foi isolada e a solução purificada em filtro de seringa de celulose regenerada (Sartoriuns Stedim, Alemanha).
2.3.4. Linearidade
A linearidade foi determinada pela curva analítica, igual a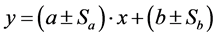 . Para construção da curva prepararam-se 10 soluções com diferentes concentrações: 0.1; 0.5; 1.0; 5.0; 50.0; 100.0; 250.0; 500.0; 750.0 e 1000.0 μg∙L−1 [10] [11] .
. Para construção da curva prepararam-se 10 soluções com diferentes concentrações: 0.1; 0.5; 1.0; 5.0; 50.0; 100.0; 250.0; 500.0; 750.0 e 1000.0 μg∙L−1 [10] [11] .
2.3.5. Faixa De Trabalho e Faixa Linear
A faixa de trabalho compreendeu o intervalo entre a menor e a maior concentração determinada pelo método via análise da linearidade.
As faixas lineares foram os intervalos contidos na faixa de trabalho que corresponderam a uma tendência linear única, além de apresentar parâmetros lineares melhores que os da curva analítica atribuída a faixa de trabalho. Ou seja, são adotadas as faixas lineares se estas apresentam curvas analíticas com valores mais adequados de r, LOD e LOQ para o método analítico, se comparados aos mesmos parâmetros para a faixa de trabalho.
2.3.6. Limite De Detecção E De Quantificação
Os limites de detecção e quantificação (LOD e LOQ) foram calculados a partir dos parâmetros lineares da curva analítica. Para isso, utilizaram-se as seguintes equações:
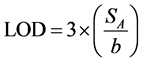
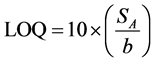
em que: Sa é o desvio-padrão de a (coeficiente angular) e b é o intercepto (coeficiente linear) [10] [11] .
2.3.7. Sensibilidade
A sensibilidade do método foi determinada utilizando a equação da reta obtida para a faixa de trabalho e, caso ocorra, para as faixas lineares.
Segundo orientação do INMETRO, a sensibilidade é obtida pelo coeficiente angular da curva analítica (termo a da equação da reta) [10] .
2.3.8. Recuperação Do Método―Exatidão
Os ensaios para recuperação foram realizados de acordo com as orientações do INMETRO. A metodologia utilizada na determinação da recuperação é a mesma descrita pela ANVISA para determinação da exatidão. Segundo o INMETRO, a exatidão deve ser avaliada numericamente por meio da recuperação.
Para verificar a recuperação/exatidão foram adicionados 10.0 mL de solução do herbicida em 1.0 g de sedimento. Foram realizados ensaios com amostras fortificadas, seguindo as recomendações da ANVISA, avaliando 3 concentrações (baixa, média e alta) com 3 réplicas cada. As concentrações do herbicida analisadas foram: 10.0, 100.0 e 1000.0 μg∙L−1.
Por meio das áreas obtidas, calculou-se a recuperação/exatidão do método:
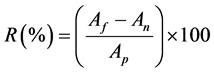
em que: R(%) é o percentual de recuperação, Af é a área do pico cromatográfico da amostra fortificada, An é a área do pico cromatográfico da amostra não fortificada e Ap é a área do pico cromatográfico do padrão analítico [10] [11] .
2.3.9. Precisão
A precisão foi analisada com base nos resultados obtidos na análise da recuperação do método. De acordo com o INMETRO e a ANVISA, a precisão deve ser avaliada pelo coeficiente de variação (CV), calculado pela equação abaixo apresentada.
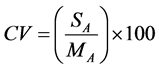
em que: CV é o coeficiente de variação, MA é a média das áreas dos picos cromatográficos obtidos e SA é o seu respectivo desvio-padrão [10] [11] .
2.3.10. Robustez
Conforme orientado pelo INMETRO, a robustez do método foi determinada via teste estatístico de Youden. Os parâmetros analisados são referentes à fase móvel e foram aqueles recomendados pela ANVISA, a saber:
i. pH: 2.0 e 2.2;
ii. Composição: 60 e 58% de acetonitrile;
iii. Fluxo: 1.4 e 1.2 mL∙min−1;
O pH foi ajustado utilizando ácido fosfórico (H3PO4) 85% (Qhemis, Brasil).Nas combinações ensaiadas para resolução do teste de Youden (Table 1), os valores utilizados na validação do método receberam uma letra maiúscula como código de identificação (A, B e C). Os valores referentes às pequenas variações receberam como código uma letra minúscula (a, b e c). Cada combinação (numerada de 1 a 8) recebeu uma letra para identificação dos resultados (de s a z).
O teste de Youden permitiu analisar o efeito de cada parâmetro, isoladamente, na robustez do método. Nas equações abaixo são apresentados os cálculos pertinentes. Os cálculos para o efeito do pH, da composição da fase móvel e do fluxo da fase móvel na robustez do método foram realizados de acordo com as seguintes equações:
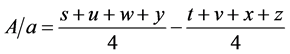
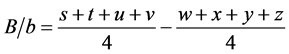
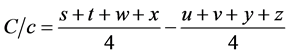
em que: A/a, B/b e C/c são os efeitos do pH, da composição da fase móvel e do fluxo da fase móvel, respectivamente, na robustez do método. As letras de s a z identificam os resultados obtidos em cada ensaio [10] [11] .
2.4. Tratamento Dos Dados e Análise Estatística
Todas as análises químicas e determinações cromatográficas foram realizadas em triplicata. Os resultados quantitativos foram apresentados na forma de média mais ou menos o erro absoluto, calculado pela equação:
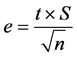
em que: e é o erro absoluto, t é o valor de t de Student com 95% de confiabilidade (P < 0.01) e 2 graus de liberdade, S é o valor do desvio-padrão e n é o número de repetições [13] .
No caso das curvas analíticas o erro foi apresentado na forma de desvio-padrão. Os cromatogramas e espectros foram apresentados após tratamento no software Microlab® Origin® versão 8.
Quando ocorrem comparações entre resultados, utilizou-se como ferramenta estatística o teste t de Student com 99% de confiabilidade (P < 0.01) e 4 graus de liberdade [13] .
3. Resultados E Discussão
3.1. Caracterização Dos Sedimentos
Analisando as características dos sedimentos (Table 2), é possível observar uma variação das características das amostras em função do uso dado ao solo.

Table 1. Combinações ensaiadas para teste estatístico de Youden, utilizado na determinação da robustez do método cromatográfico.
Table 2. Resultados da caracterização química e física das amostras de sedimentos.
Resultados na mesma linha se uidos pela mesma letra na o apresentam diferenc as significativas de acordo com teste t de studentem P < 0.01.
Comparando os teores de matéria orgânica (MO), os sedimentos da amostra Zona Pecuária apresentaram diferença significativa se comparados às demais amostras (teste t de Student, P < 0.01 e 4 graus de liberdade): aproximadamente 60% de diferença em comparação à amostra Zona Agrícola e 100% para amostra Estação Ecológica.
A amostra Estação Ecológica apresentou um teor menor de matéria orgânica (8.37%), porém esta diferença é insignificante se comparado ao teor da amostra Zona Agrícola (10.72%). Dentre as funções da matéria orgânica está a capacidade de retenção de compostos orgânicos nos solos e sedimentos, o que pode interferir, principalmente, na recuperação do método.
Analisando os teores dos ácidos húmicos, nenhuma amostra apresentou diferença significativa na comparação entre as três amostras de sedimento analisadas (teste t de Student, P < 0.01 e 4 graus de liberdade).
Analisando os resultados da capacidade de troca catiônica (CTC), todas as amostras apresentaram diferenças significativas (teste t de Student, P < 0.01 e 4 graus de liberdade). Na comparação entre as amostras Zona Pecuária e as demais os valores obtidos foram bastante elevados, aproximadamente 100% de diferença em comparação a amostra Zona Agrícola e 300% para amostra Estação Ecológica. Os sedimentos da Estação Ecológica apresentaram menor CTC (24.87 cmolc kg−1). A capacidade de troca catiônica é um parâmetro relevante que pode influenciar na adsorção dos compostos orgânicos. Quanto maior a quantidade de compostos adsorvidos, menor quantidade permanecerá livre em solução, o que poderá dificultar a determinação, influenciando na validação do método [16] [17] .
A classe textural dos sedimentos foi determinada via análise granulométrica. Os sedimentos analisados apresentaram diferentes percentuais de grãos. As amostras Zona Agrícola e Estação Ecológica apresentaram um maior teor de areia (55 e 75.7%, respectivamente). A amostra Zona Pecuária foi a que apresentou um maior percentual de argila (35%) e silte (45.20%). O diâmetro das partículas está relacionado a sua área superficial. Partículas menores (por exemplo, argila e silte) possuem maior área superficial e, com isso, a retenção de compostos orgânicos e inorgânicos é mais intensa. Uma vez retidos, os compostos podem sofrer transformação (oxidação ou redução), interferindo em sua detecção, prejudicando o ensaio que se deseja realizar [18] [19] .
3.2. Validação Do Método Cromatográfico
3.2.1. Seletividade
Os ensaios para obtenção da seletividade do método iniciaram-se com o registro do espectro na região do ultravioleta-visível (Figure 3). O comprimento de onda de 235 nm foi o que apresentou maior absorção da radiação, logo este valor foi utilizado na calibração do detector UV do cromatógrafo.

Figure 3. Espectro de UV-Vis do herbicida etilclorimuron 1 mg∙L−1.
Analisou-se a presença de picos cromatográficos com tempos iguais nos cromatogramas do padrão analítico (herbicida etilclorimuron) (Figure 4) e dos três sedimentos em análise (Figure 5). Observando os resultados obtidos, a retenção do analito deu-se até os 2.91 minutos da determinação cromatográfica. É possível observar que, no tempo de retenção do analito de interesse, não foram detectados outros picos cromatográficos com tempos de retenção coincidentes entre a matriz e o herbicida etilclorimuron. A composição dos sedimentos não apresentou influência na seletividade do método. O fluxo elevado (1.4 mL∙min−1) juntamente com a alta polaridade da fase móvel permitiu a aglomeração dos picos cromatográficos referentes à matriz analítica nos primeiros instantes da separação cromatográfica, permitindo a detecção do herbicida sem interferentes. Assim, garante-se a seletividade do método [7] .
3.2.2. Linearidade
Para determinação da linearidade, utilizaram-se soluções do herbicida com concentrações variando de 0.1 a 1000.0 μg∙L−1. Os picos cromatográficos só foram obtidos e quantificados a partir de 5.0 μg∙L−1. Neste sentido, a faixa de trabalho atendida pelo método abrangeu o intervalo entre 5.0 e 1000.0 μg∙L−1.
Table 3 apresenta as curvas analíticas e seus parâmetros lineares para a faixa de trabalho (FLi) e duas faixas lineares (FLiiA e FLiiB) (Table 3).
A faixa de trabalho (FLi) correspondeu ao intervalo entre a menor concentração determinada pelo método e a maior analisada (5.0 a 1000.0 μg∙L−1). A primeira faixa linear atendeu ao intervalo de 5.0 a 250.0 μg∙L−1 (FLiiA) e a segunda faixa de 250.0 a 1000.0 μg∙L−1 (FLiiB).
Em função dos valores mais adequados de r, LOD e LOQ, optou-se por trabalhar com duas faixas lineares. A escolha da equação da reta para os cálculo da concentração deu-se pela área do pico cromatográfico. Picos com área de até 20,324.33 foramtratadas por meio da equação da reta da faixa linear FLiiA. Acima deste valor deve-se a equação da reta referente à faixa linear FLiiB.
Não foram calculados os valores de sensibilidade, LOD e LOQ para a faixa linear FLiiB pois as concentrações analisadas não se encontram no limite inferior da curva analítica, aos quais estes parâmetros são inerentes.
Analisando os coeficientes de correlação linear (r), todos os resultados estão de acordo com os valores orientados. Um coeficiente de correlação maior que 0.999 é considerado uma evidência de um ajuste ideal dos dados para uma regressão linear. Para a ANVISA, a correlação mínima aceitável deve ser igual ou superior a 0.99. Para o INMETRO esse valor é de 0.9 [10] [11] .

Figure 4. Cromatograma do herbicida etilclorimuron 1.0 mg∙L−1 obtido por HPLC-UV.

Figure 5. Cromatogramas dos extratos dos sedimentos em acetonitrila obtidos por HPLC-UV.
Table 3. Resultados obtidos na validação do método cromatográfico para os parâmetros: linearidade, sensibilidade, limites de detecção e de quantificação.
3.2.3. Sensibilidade
Analisando o coeficiente de variação angular (valor de a da curva analítica) das retas obtidas (Table 3), a faixa linear FLiiA é a que apresenta uma maior sensibilidade. Comparando com a faixa linear FLi, este valor é bastante significativo. A diferença na sensibilidade entre estas duas faixas lineares é de aproximadamente 41.74%. O INMETRO não orienta um valor para a sensibilidade do método.
Considerando ser relevante uma sensibilidade alta para as concentrações baixas (mais difíceis de serem detectadas) é justificável a utilização de duas faixas lineares ao invés de apenas uma.
3.2.4. Limite De Detecção (LOD) e Limite de Quantificação (LOQ)
Os limites de detecção (LOD) e de quantificação (LOQ) foram determinados via curva analítica, considerando as duas faixas lineares. Analisando os valores obtidos (Table 3), os valores de LOD e LOQ são significativamente inferiores quando a faixa de trabalho é dividida em duas faixas lineares.
O valor do LOD da faixa FLiiA é aproximadamente 575% menor que aquele limite em FLi. Para o LOQ esta diferença é de 517%. Esta considerável diferença é mais uma evidência para a utilização de duas faixas lineares. Nestas análises, a faixa linear FLiiB foi desconsiderada por não contemplar as concentrações mais baixas distribuídas ao longo da faixa de trabalho.
Os documentos do INMETRO e da ANVISA não contemplam um valor a se validar para estes parâmetros. Tratando-se de pesticidas ou outras moléculas com ação danosa ao homem ou ao meio ambiente, são as agências e autarquias públicas de regulamentação ambientais que determinam estes parâmetros como Valor Máximo Permitido (VMP), e cabe aos métodos analíticos validarem seus LOD e LOQ atendendo a esta exigência, normalmente imposta por órgãos e autarquias ambientais. No caso do etilclorimuron, não existem VMPs associado a este herbicida.
3.2.5. Recuperação Do méTodo―Exatidão
Os ensaios de recuperação foram realizados a fim de contemplar a recomendação do INMETRO. Os valores obtidos (Table 4) indicaram que, quanto maior a concentração analítica analisada, menor foi o efeito da matriz. Isso fez com que a recuperação fosse maior nos ensaios com concentrações elevadas.
Considerando que a recuperação é um ensaio quantitativo, a concentração analítica de 10.0 μg∙L−1 encontra-se abaixo do LOQ (29.48 μg∙L−1), logo se justifica a dificuldade do método em obter um percentual de recuperação considerável, próximo a 100%.
Comparando os percentuais de recuperação obtidos e seus respectivos erros absolutos, houve diferença significativa apenas ao comparar as concentrações analíticas analisadas em 1000.0 e 100.0 μg∙L−1 na amostra Zona Agrícola (aproximadamente 72% de diferença). Nas demais amostras analisadas, tais comparações não excederam 20% de diferença. Esta diferença pode estar sendo influenciada pela composição química dos sedimentos. O INMETRO não contempla um valor orientativo para os ensaios de recuperação.
Table 4. Resultados obtidos na validação do método cromatográfico para os parâmetros: recuperação-exatidão e precisão.
Mesmo possuindo composições diferentes, os sedimentos influenciaram pouco na recuperação. Como o tempo de contato foi de 30 minutos, possivelmente não ocorreram processos sortivos ou de degradação significativos, a ponto de interferir nos resultados.
De acordo com a ANVISA, um método é considerado exato se este possuir uma recuperação entre 80 e 120% da concentração analítica inicial. O método proposto atendeu à esta consideração em quase todas as situações (Table 5). Os valores obtidos para a concentração de 10.0 μg∙L−1 não foram validados pois apresentaram valores abaixo de 80%, valor mínimo aceitável. Além disso, a concentração de 10.0 μg∙L−1 está abaixo do limite de quantificação, justificando a dificuldade do método em quantificar o analito nestas condições.
A amostra Zona Agrícola, na concentração de 1000.0 μg∙L−1, apresentou uma recuperação de 133.33%, aproximadamente 13% acima do permitido. A dificuldade do método em recuperar a concentração exata pode- estar relacionada a superdosagem do herbicida etilclorimuron, a qual pode causar uma pertubação no sistema e detecção cromatográfica.
3.2.6. Precisão
A precisão foi calculada a partir dos resultados obtidos na recuperação do método (Table 4), por meio do cálculo do coeficiente de variação. De acordo com as recomendações da ANVISA, não são admitidos coeficientes de variação acima de 15%. Para o INMETRO, os coeficientes de variação máximos permitidos são dados em função de cada concentração analisada. Para as concentrações de 1000.0, 100.0 e 10.0 μg∙L−1 estes valores são 16, 23 e 32%, respectivamente. Assim, os valores obtidos foram validados de acordo com as recomendações da ANVISA e do INMETRO, para todas as amostras de sedimentos e em todas as concentrações estudadas.
3.2.7. Robustez
A robustez do método foi analisada aplicando pequenas alterações nos parâmetros cromatográficos em validação. Estas variações implicaram em alterações nas áreas dos picos cromatográficos e no tempo de retenção do herbicida etilclorimuron. Devido à relação linear que existe entre as áreas dos picos e a concentração analítica, a concentração inicial (100.0 μg∙L−1) foi quantificada com diferentes valores.
Analisando os resultados obtidos (Table 5), a diminuição no fluxo da fase móvel causou um acréscimo de aproximadamente 1 minuto em quase todos os ensaios. O aumento no tempo de retenção foi ainda mais significativo nas combinações em que ocorreu a diminuição no percentual de solvente na fase móvel. Possivelmente o aumento do percentual da solução (H2O + H3PO4 0.1%) na composição da fase móvel aumentou a interação entre o analito e a fase estacionária da coluna cromatográfica, aumentando a solubilidade do analito no leito da coluna cromatográfica, fazendo com que este ficasse retido por mais tempo [7] .
As variações de pH interferiram com menor intensidade na detecção do herbicida, possivelmente relacionado à sua alta solubilidade em solvente orgânico.
Aos valores apresentados na Table 5 aplicou-se o teste de Youden para o cálculo da robustez. O método mostrou-se mais robusto para as pequenas variações do pH da solução que compõem a fase móvel (A/a = 0.92), alterando irrisoriamente os resultados, seguido pelo fluxo (C/c = 7.20) e, por último, a composição da fase móvel (B/b = 14.86).
Table 5. Resultados obtidos na validação do método cromatográfico para o parâmetro robustez.
4. Conclusões
Nos parâmetros e condições descritas, o método cromatográfico para a determinação do herbicida etilclorimuron em sedimentos mostrou-se eficaz. Os parâmetros cromatográficos validados permitirama determinação do analito de interesse com pouca interferência da matriz. Os resultados foram validados para a linearidade no intervalo de 5.0 a 1000.0 μg∙L−1, com boa sensibilidade e baixos LOD e LOQ. Além disso, o método mostrou-se robusto para os parâmetros analisados: pH, composição e fluxo, todos relacionados à fase móvel. Nos ensaios de recuperação com concentrações acima do LOQ, os valores obtidos foram quase sempre exatos e precisos, com pouca interferência da matriz.
Assim, considera-se o método cromatográfico proposto adequado para análise do herbicida etilclorimuron em sedimentos, uma vez que se adequou aos documentos normativos e orientativos do INMETRO e ANVISA e, quando cabível de valores orientativos, os resultados obtidos se encontram em concordância.
Agradecimentos
À Coordenação de Aperfeiçoamento de Pessoal de Nível Superior (CAPES) pela bolsa concedida a Nunes R.R., ao Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq)pelo apoio financeiro e ao professor Dr. Luís César Schiesari (EACH-USP) por ceder as amostras de sedimentos utilizadas neste estudo.
Cite this paper
Ramom Rachide Nunes,Bruno Henrique Froes Santos,Maria Olímpia de Oliveira Rezende, (2016) Analytical Procedures and Method Validation for Determination of Chlorimuron Ethyl in Sediments. Open Access Library Journal,03,1-12. doi: 10.4236/oalib.1102398
References
- 1. Baird, C. (2008) Environmental Chemistry. Freeman, New York.
- 2. US-EPA—United States Environmental Protection Agency (2005) Citizen’s Guide to Pest Control and Pesticide Safety. EPA, Washington.
- 3. Oliveira Junior, R.S., Constantin, J. and Inoue, M.H. (2011) Biologia e manejo de plantas daninhas. Omnipax, Curitiba.
- 4. Hay, J.V. (1990) Chemistry of Sulfonylurea Herbicides. Pesticide Science, 29, 247-261.
http://dx.doi.org/10.1002/ps.2780290303 - 5. ANVISA—Agência Nacional de Vigilancia Sanitária (1987) Monografias técnicas do ministério da saúde sobre substancias com ação tóxica sobre animais ou plantas, Portaria 9 10/85. ANVISA, Brasília.
- 6. Zanella, R., Primel, E.G., Gonç;alves, F.F., Kurz, M.H.S. and Mistura, C.M. (2003) Development and Validation of a High-Performance Liquid Chromatographic Procedure for the Determination of Herbicide Residues in Surface and Agriculture Waters. Journal of Separation Science, 26, 935-938.
http://dx.doi.org/10.1002/jssc.200301309 - 7. Lanças, F.M. (2009) Cromatografia liquida moderna—HPLC/CLAE. RiMa, São Carlos.
- 8. Leite, F. (2008) Validação em análise química. Átomo, Campinas.
- 9. Lanças, F.M. (2004) Validação de métodos cromatográficos de análise. RiMa, São Carlos.
- 10. INMETRO—Instituto Nacional de Metrologia, Normalização e Qualidade Industrial (2011) Orientaçõ es sobre Validação de Métodos de Ensaios Químicos, DOQ-CGCRE-008. INMETRO: Brasília.
- 11. ANVISA—Agência Nacional de Vigilancia Sanitária (2003) Guia para validação de métodos analíticos e bioanalíticos, Resolução RE n° 899. ANVISA, Brasília.
- 12. ISO—International Standard Organization (1999) General Requirements for the Competence of Testing and Calibration Laboratories, ISO/IEC 17025. ISO, Genebra.
- 13. Johnson, R.A. and Bhattacharyya, G.K. (2001) Statistics Principle and Methods. John Wiley, New York.
- 14. ANA—Agencial Nacional de Águas, CETESB—Companhia Ambiental do Estado de São Paulo (2011) Guia Nacional de Coleta e Preservação de Amostras: Água, Sedimentos, Comunidades Aquáticas e Efluentes Líquidos. ANA, Brasília.
- 15. Nogueira, A.R.A. and Souza, G.B. (2005) Manual de laboratòrios: solo, água, nutrição vegetal, nutrição animal e alimentos. EMBRAPA Pecuária Sudeste, São Carlos.
- 16. Hayes, M.H.B., MacCarthy, P., Malcolm, R.L. and Swift, R.S. (1989) Humic Substances II: In Search of Structure. John Wiley, New York.
- 17. Carroll, D. (1959) Ion Exchange in Clays and Other Minerals. Geological Society of America Bulletin, 70, 749-779.
http://dx.doi.org/10.1130/0016-7606(1959)70[749:IEICAO]2.0.CO;2 - 18. Tan, K.H. (1998) Principles of Soil Chemistry. Marcel Dekker, New York.
- 19. Essington, M.E. (2004) Soil and Water Chemistry: An Integrative Approach. CRC Press, Boca Raton.
NOTES

*Corresponding author.