Open Access Library Journal
Vol.02 No.06(2015), Article ID:68447,10 pages
10.4236/oalib.1101601
Simulations Relating to the Determination of Protein Secondary Structure Fractions from Circular Dichroism Spectra
David A. Haner, Patrick W. Mobley*
Department of Chemistry and Biochemistry, California State Polytechnic University, Pomona, USA
Email: *pwmobley@cpp.edu
Copyright © 2015 by authors and OALib.
This work is licensed under the Creative Commons Attribution International License (CC BY).
http://creativecommons.org/licenses/by/4.0/



Received 26 May 2015; accepted 11 June 2015; published 18 June 2015
ABSTRACT
Test problem simulations are presented of the matrix equation, D = BF, equivalent to least squares data fitting, where the matrices are rectangular with D and F being experimental data. The chosen application is finding fractions of secondary structures of proteins from circular dichroism (CD) spectra employing singular value decomposition, SVD, to obtain the matrix B and its pseudo- reciprocal. In practice the first step of the analysis is to select the reduced noise representation of the CD spectra which sets the bounds for subsequent computation and the development of the CD noise spectra. The conclusion from analysis is to obtain the structural component spectra summarizing the database and all the structural fractions and their uncertainties. The database noise spectrum can be used to prepare the reduced noise CD spectrum for a new protein to yield its structural fractions and their uncertainties.
Keywords:
Singular Value Decomposition, Circular Dichroism, Protein Structure
Subject Areas: Computational Biology, Numerical Methods

1. Introduction
Mathematical modeling of scientific data is at the foundation of modern scientific exploration of natural phenomena. Usually a model of the phenomenon is hypothesized and translated into quantifiable variables and compared with the related laboratory measurements [1] - [6] . The analysis of circular dichroism (CD) spectra of proteins for their secondary structure content is a typical example of modern mathematical modeling of laboratory data to quantify molecular structure [7] - [10] . This effort has been under development for several decades and is still evolving today due to technological advances and ever increasing progress into the understanding of biochemical processes.
Initially the problem was attacked using the techniques that were current, namely constructing the spectral functions describing secondary structures presumed to be present and responsible for the CD spectra. The quality and quantity of the CD spectra has improved with each passing year; however the care required in obtaining high quality experimental data has not lessened. To maximize the usefulness of experimental data, not only should each laboratory take great care in obtaining it, but a generally acceptable protocol should be employed to ensure that the quality of the data is easily documented. The results from the analysis of the quality data are made credible by presenting output obtained by the treatment of selective test problems with the analytical algorithms. The protocol must be comprehensive enough to ensure reproducible measurement results of reproduced materials and environments. It is then that confidence can be drawn from the implication of the analytical results. In short, there is no substitute for precise data. After the necessary database is complete with calibrated and validated data, then the analytical data reduction into model parameters follows. One of the first steps in the development or testing of analytical modeling is the design of test problems. Adequate test problems are those which simulate the real database and demonstrate or quantify the prediction capability of the algorithms employed.
Deriving protein secondary structure content from CD spectra has changed from postulating component spectra of the characteristic structures to the use of the secondary structure fractions from X-ray data as a basis for the mathematical prediction of the component spectra representation of the CD database. The mathematical technique used is well developed [3] and is usually found under the heading “Factor Analysis.” The CD spectra of proteins are digitized intensity signals taken over intervals of the optical spectrum and the secondary structure fractions are derived from X-ray measurements for each protein included in the database [7] . This information is presented as arrays of numbers or matrices, usually rectangular. Usually the CD and X-ray data are analyzed employing the techniques of singular value decomposition, SVD [2] [6] . In fact, current programs that are widely used in analyses of CD data, Dichroweb [11] and CDPro [12] , are largely based on SVD as described by Forsythe, et al. [2] with various restrictions to the database and the acceptability of the results. (The Dichroweb site has good reviews of CD spectra analysis techniques on the home page and under Background Information/ About Deconvolution Algorithms.) Rarely are the fractions of secondary structures reported with the concomitant standard deviations or is the experimental error associated with the resulting functional fitting to the CD spectrum available. This omission is discussed in the analysis presented in the Results and Discussion section.
1.1. Theory
Simulation of a solution strategy helps to validate its use and to define its limitations and instability. The solution strategy in operation here is to start with two independent experimental measurements on a group of proteins and to combine these measurements to uncover fundamental structural factors that are common to all the proteins. The two experimental measured quantities used in this case are the fractions of secondary structures for each protein as measured by X-ray crystallography and their individual calibrated CD spectrum. Thus,
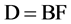 (1a)
(1a)
where:
D = CD spectra (calibrated);
B = the basis function (fundamental factors), to be determined;
F = the secondary structure fractions (X-ray results).
Generally, the variables are rectangular matrices reflecting that the CD spectrum for each protein is a digital listing and the number of proteins must be a number greater than or equal to the number of secondary structures fractions. The rank of the CD data, D, must agree with the rank of the fraction data, F, and is achieved by the number of singular values retained. The resulting data matrix is the reduced matrix, D+. Then
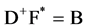 (1b)
(1b)
where F* is the pseudo-inverse of F.
This problem can be reformulated from a different point of view, though related to the first. That is,
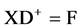 (2a)
(2a)
where
X = projector functions (a.k.a.: generalized inverse functions [7] );
D+ = CD spectra (calibrated), reduced;
F = the fractions (X-ray results).
And
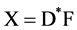 (2b)
(2b)
where D* is the pseudo-inverse of D+.
The relationship between the two points of view is that X and B are pseudo-reciprocal to each other. Equation (1) can be used to find the basis functions, B, which characterize the connection between matrices D+ and F. Once B is defined it will be used with other spectra of proteins to find corresponding values of the secondary structure fractions and their confidence intervals or uncertainties. Correspondingly, Equation (2) could be solved to obtain the matrix X to project the fractional values from the spectrum of a protein from the reduced database. By finding B, the pseudo-reciprocal of X, the solution by Equation (1) leads directly to finding the confidence intervals for the fractions obtained by function fitting to the reduced CD data.
There is another equivalent point of view for solution of this model. Transforming to physically meaningful representation from the SVD formulation [2] [6] : factorization of rectangular matrices.
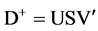 (3a)
(3a)
where
U = column orthogonal matrix;
S = diagonal matrix of singular values;
V' = row orthogonal matrix, transpose.
Introducing T, the rotation matrix
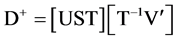 (3b)
(3b)
equating the factors to the experimental factors; thus
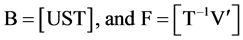
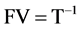 (3c)
(3c)
and further T and then B.
The algorithms of SVD set the dimensions of the matrices after reducing the rank by choosing the number of singular values retained to the same number as the primary factors (secondary structure fractions). Since generally the matrices are rectangular, and therefore the subsequent factors are rectangular matrices, the techniques of finding pseudo-inverses are required [3] . In order to accommodate the numerical solutions of the two parts of the protein database, digital programs will be written, compiled, and executed as the solution develops. Part of the program development is to use test problems to prove results from the algorithms employed. An efficient use of test problems is to design them so as to finally bring the test solution to mimic a simulation of a realistic problem.
1.2. Simulations
To begin the simulations of this model one must choose the number of basis functions, the size of the spectral range of the database, the number of proteins, and their fractions of secondary structures. In order to validate and perfect the digital algorithms involved in the solution of the protein databases, it is valuable to formulate a simulation of the CD spectral portion of the database by selecting the basis functions possessing some of the characteristics for the structure. This is achieved by choosing well-defined, distinct functions defined over the same wavelength range anticipated for the laboratory setting. In this application 81 points in the wavelength interval for 20 spectra with four secondary structures were chosen.
The four basis functions chosen relate to the first four harmonic oscillator functions (SHM), which have some orthogonality character (that is orthogonality on + to − infinity, a one dimensional domain) though not complete on the finite domain of digital functions as shown in Figure 1. Those functions are used with the specially designed secondary fractions matrix that sum to one for the possible structure fractions and that are uniformly distributed over the group of simulated proteins, so that the list of fractions of each structural type over the protein group sum to the same number. That is the sum of all the entries of the rows sum to the same number, one; and such that all the entries in the columns summed yield the same unique number as shown in Table 1.
After the matrix, D is constructed using the functions, B, as prescribed and proportioned by the matrix, F, then the algorithms of the solution are applied to matrices, D and F to obtain the matrices, B and X corresponding to Equations (1)-(3). The solution algorithms should yield the basis functions, B that are pseudo-reciprocal to X, the projector functions. The results of the simulations should yield the known basis functions and a least squares fit to the spectra should yield the elements of the fractions matrix, F, and provide precision indices or standard

Figure 1. Harmonic oscillator basis functions. The four orthogonal basis functions, B, used to synthesize the simulated spectra from the fractions of secondary structures of Table 1. Simple harmonic motion (SHM) function 1: blue diamond; SHM2: brown square; SHM3: green triangle; SHM4: purple circle.

Table 1. Secondary structure fractions for a database of virtual proteins. The secondary structures fraction, F, a rectangular matrix, has columns: I, II, III, and IV, which represent four types of secondary structures. For real proteins these structures are often from X-ray studies and typically would be the canonical examples: α-helix, β-structure, etc. Each row lists the fractions of the structures present for that protein. Each row sums to one, and each column (proteins 1-20) sums to the same number.
deviations for the fractions on the constructed database. The solution for the projectors when applied to the spectra should yield the corresponding structural fractions of the spectrum. Further, X and B should prove to be pseudo-reciprocal.
2. Materials and Methods
This type of scientific modeling can be developed on digital computers using any of many high-level computational compilers. We used Microsoft Fortran Powerstation Professional Development System, Version 1.0 for MS-DOS and Windows Operating Systems, 1993, U.S. patent No. 4,955,066, running on a Dell Dimension 2400 PC. Various subroutines were taken directly from references 1, 2, and 6 (some of these subroutines are available in C in more recent publications by these last authors), while the main programs were our adaptations written in the Fortran Powerstation above (very similar to Fortran 77). SVD was checked by running the equivalent generalized matrix inversion. The criteria for the selection of the virtual secondary structure fractions are described in the text and the legend of Table 1. The data type was generally double precision.
3. Results and Discussion
The simulation database, D, is a matrix of 81 rows and 20 columns. Each column represents the CD spectrum of a virtual protein. The first step in the solution is to obtain the singular values for the data matrix, D. The results are shown in Table 2, and reflect the characteristics of the simulation. Also shown in Table 2 are the residual standard deviation and the variance which are directly related to the singular values (see reference 3 for details). Addition of the variances can be used to estimate the importance of each singular value and the order of the reduction. Since the utilization of archived protein data has two independent parts, the CD spectra and the X-ray derived secondary structure fractions, each part should be analyzed for singular values to insure that the rank of the independent parts are in agreement.
Table 3 shows the singular values from SVD of the matrix, F, Table 1. The SVD algorithm used in our analysis does not order the singular values, however the largest values are chosen due to their importance. The fact that the singular values and variances are large and similar is due to the details of the simulation; SVD of actual database components will probably be more varied. The next step in the algorithm is to find the basis functions by Equations (1)-(3), and the pseudo-reciprocal projectors. The solution basis functions are duplicates of the starting basis functions shown in Figure 1. The solutions for the pseudo-reciprocal projectors are shown in Figure 2. These functions are nearly identical to the basis functions except for a change in amplitude. This is to be expected since orthogonal functions are pseudo-reciprocal.
Table 2. SVD results for SHM database.
Table 3. SVD result for fractions of Table 1.

Figure 2. Pseudo-reciprocal functions for SHM. Functions are reciprocal to those of Figure 1. From SHM#: 1―diamond, 2―square, 3―triangle, 4―circle.
Application of the pseudo-reciprocal basis functions (projectors) to the simulated CD spectra showed the secondary structure fractions for each virtual protein of Table 1. To further confirm the reciprocity of the projectors to the basis functions, a least squares fit of the computed basis functions to virtual protein number 5, shown in Figure 3, was completed. The resulting fractions were identical; the fit function is not shown as it is identical to within machine precision. The results from the least squares fitting (LSQ) of spectrum no 5 using the computed basis functions are shown in Table 4. The fractions are the linear coefficients of the LSQ for the basis functions to the reduced CD spectrum. The uncertainty in the secondary fractions are the product of the deviation factors times the sample variance [1] . The uncertainties in parameters for real data should be larger regardless of the precision of the computation.
To further test simulations to the solution algorithms a linearly independent (LID) set of basis functions [6] , shown in Figure 4, were chosen to construct the virtual protein CD spectra database using the secondary structure fractions of Table 1. The singular value decomposition of the simulated CD data was computed and the results are shown in Table 5. These results show that the singular values reflect information about the independence of the functions and fractions. The simulation has some features that mimic a real CD database such as decreasing singular values. The functions, shown in Figure 5, are clearly not the same shape as those in Figure 4, demonstrating the lack of orthogonality.
Application of these functions to the simulated spectra showed the structure fractions for each virtual protein as given in Table 1. The spectrum of virtual protein number 5 is shown in Figure 6. This spectrum does not resemble the spectrum shown in Figure 3; however the fit parameters are identical as they should be with variances within machine precision. A summary of the linear least squares fitting of virtual protein number 5 is shown in Table 6.
In summary, the results are as follows: The pseudo-reciprocals were obtained using the two algorithms, SVD and general matrix methods. The results are duplicated to within anticipated machine precision at double precision and the standard deviation calculated for the solution functions arrived at by the different approaches show the high degree of agreement again governed by machine precision. The singular values obtained for the decomposition of the D and F matrices indicate well defined completeness of the simulations components and the adequacy of the algorithms used in the digital programs. In developing the simulations and the cross checking nature of the solution strategy, the programming of the algorithms was maximized.
4. Conclusions
Any of the three formulations presented for finding the basis functions and/or projector functions, Equations (1)-(3), are adequate when employed on precisely generated test problems. Testing on precise data requires no choices to be made in regard to the number of singular values to be retained in view of the variance obtained for

Figure 3. Simulated spectrum for protein #5. The simulated spectrum of virtual protein number 5 (see Table 1) and its resulting computed fit superimposed.
Table 4. Results for LSQ of virtual protein #5. I through IV are secondary structures.
S, sample variance = 0.200E−14.
Table 5. SVD result for LID database LID: linearly independent basis functions.
Table 6. Results for LSQ of virtual protein #5 for LID basis set. I through IV are secondary structures.
S = sample variance = 0.585E−14.

Figure 4. Linearly independent basis functions (LID). All ranges of the independent variable are chosen to fit within the simulation range. Function (Fn) 1: Gaussian-blue diamond; Fn 2: Sine-brown square; Fn 3: Bessel of zero order- green triangle; Fn 4: Newman’s Bessel of second kind of zero order-purple circular dot.

Figure 5. Pseudo-reciprocal functions for LID Functions are reciprocal to those of Figure 4 and show the lack of orthogonality between the two groups. Function (Fn) 1: Pseudo-reciprocal (PsR) of Gaussian-blue diamond; Fn 2: PsR of Sine-brown square; Fn 3: PsR of Bessel of zero order-green triangle; Fn 4: PsR of Newman’s Bessel of second kind of zero order-purple circular dot.
the CD data or the X-ray secondary structure fractions as the number of singular values is set by the test problem. If one has access to and understanding of the computer code, then one can insert extra steps to produce results of other checks to the veracity of the analysis program. If not, then inputting designed virtual data and obtaining reasonable output will help to build confidence in the numerical solution.
When processing real laboratory data the choice of the number of singular values retained is determined by the number of secondary structures expressed in the fractions data. The remaining choices in the steps to obtain

Figure 6. Protein number 5 spectrum for LID Spectrum of protein number 5 computed and fitted traces are identical.
the basis functions can be determined by choosing the set of functions that yield the smallest standard deviation of the function fitting of the reduced CD spectrum or spectra. Finally the old adage “garbage in, garbage out” prevails in the digital age. Thus high quality, calibrated, standardized and validated input data will give the high quality, reliable and reproducible output results.
The reward for the labor of algorithm development and precise data accumulation is realized by obtaining the basis set of functions for the database, reciprocal projectors, and the noise database. These results lead directly to finding the secondary structure fractions from the CD of a new protein. This is accomplished by using all the information available from the database and the algorithms used in the analysis. The mathematical description is

where
D = the CD database;
D+ = the reduced database for the retained singular values;
E = the noise database for the negligible singular values.
The rectangular matrix, E, the same size as D, contains all the unused information or noise present in the CD spectra and D+ contains all the useful information present in the CD spectra. Any new protein CD spectrum is composed of useful information and noise. What is desired is to remove the noise from the new spectrum and then to process the reduced spectrum by the projection or basis functions to obtain the secondary structure fractions present. This is done by finding the matrix E for the database as part of the first steps before finding the projectors, X, and the basis, B. There is a noise spectrum for each protein in the database in the matrix E and they are representative of the noise spectra of the proteins contained in the database. A simple average of the noise spectra should be subtracted from the CD spectrum of the target protein to obtain a reasonable approximation of its reduced spectrum. The reduced spectrum is then subjected to the database-derived projectors X or basis B to obtain the estimated secondary structure fractions and their uncertainties. Note that this technique works best when the database fits the criteria contained in the Introduction and the legend to Table 1: the sum of the secondary structure decimal fractions should be one for a selected protein (horizontally in Table 1) and the same number within a selected secondary structure (vertically in Table 1). The database needs to be four or more proteins; adding more does not help unless they move the set closer to the criteria.
In future studies we will obtain CD spectra and X-ray secondary structure fractions from published databases and subject selected lists of proteins to analysis as described here. We will describe the error estimation on parameters of the model in relation to the significant precision of the input data. Our preliminary studies of the computed prediction of secondary structure fractions for a reduced CD data and X-ray secondary structure database show that a single parameter adjustment of the CD spectra will rectify the ambiguity in the summation of the fractions to one per protein.
Acknowledgements
The authors thank Anne T. Tang for assistance in formatting and Dr. Lisa Alex for her support and encouragement.
Cite this paper
David A. Haner,Patrick W. Mobley, (2015) Simulations Relating to the Determination of Protein Secondary Structure Fractions from Circular Dichroism Spectra. Open Access Library Journal,02,1-10. doi: 10.4236/oalib.1101601
References
- 1. Bevington, P. (1969) Data Reduction and Error Analysis for the Physical Sciences. McGraw-Hill, New York, 154.
- 2. Forsythe, G.E., Malcolm, M.A. and Moler, C.B. (1977) Computer Methods for Mathematical Computations. Prentice-Hall, Inc., Englewood Cliffs, 192.
- 3. Malinowski, E.R. (1991) Factor Analysis in Chemistry. 2nd Edition, John Wiley and Sons, New York, 58, 85, 95, 111.
- 4. Olver, P.J. and Shakiban, C. (2006) Applied Linear Algebra. Pearson Prentice Hall, New Jersey, 425.
- 5. Perrin, C.L. (1970) Mathematics for Chemists. John Wiley & Sons Inc., New York, 247.
- 6. Press, W.H., Flannery, B.P., Teukolsky, S.A. and Vetterling, W.T. (1986) Numerical Recipes. Cambridge University Press, Cambridge, 52, 60, 172.
- 7. Compton, L.A. and Johnson, W.C. (1986) Analysis of Protein Circular Dichroism Spectra for Secondary Structure Using a Simple Matrix Multiplication. Analytical Biochemistry, 155, 155-167.
http://dx.doi.org/10.1016/0003-2697(86)90241-1 - 8. Hennessey, J.P. and Johnson, W.C. (1981) Information Content in the Circular Dichroism of Proteins. Biochemistry, 20, 1085-1094.
http://dx.doi.org/10.1021/bi00508a007 - 9. Hennessey, J.P. and Johnson, W.C. (1982) Experimental Errors and Their Effect on Analyzing Circular Dichroism Spectra of Proteins. Analytical Biochemistry, 125, 177-188.
http://dx.doi.org/10.1016/0003-2697(82)90400-6 - 10. Miles, A.J., Whitmore, L. and Wallace, B.A. (2005) Spectral Magnitude Effects on the Analyses of Secondary Structure from Circular Dichroism Spectroscopic Data. Protein Science, 14, 368-374.
http://dx.doi.org/10.1110/ps.041019905 - 11. Whitmore, L. (2001) DichroWeb—Online Circular Dichroism Analysis.
http://dichroweb.cryst.bbk.ac.uk/html/home.shtml - 12. Sreerama, N. (2004) CDPro—A Software Package for Analyzing Protein CD Spectra.
http://lamar.colostate.edu/~sreeram/CDPro/
NOTES

*Corresponding author.