 Materials Sciences and Applicatio n, 2011, 2, 1166-1174 doi:10.4236/msa.2011.28157 Published Online August 2011 (http://www.SciRP.org/journal/msa) Copyright © 2011 SciRes. MSA Theoretical Study of the Rare Earth Compounds LaFe13-xTx (T = Cr, Cu, Ga, Mn, Ni) and Curie Temperature Variation Lingping Xiao1,2*, Jiang Shen2 1Jiangxi Blue Sky University, Nanchang, China; 2Institute of Applied Physics, University of Science and Technology, Beijing, China. Email: *xiaolingping1982@163.com Received November 25th, 2010; revised January 17th, 2011; accepted June 9th, 2011. ABSTRACT The phase stability and site preference of the intermetallics LaFe13-xTx (T = Cr, Cu, Ga, Mn, Ni) with NaZn13-type struc- ture have been investigated by lattice inversion potentials. The calculated results indicate that each of the stabilizing elements Cr and Mn significantly decreases the cohesive energy of LaFe13-xTx and plays a role in stabilizing the 1:13 structure. The calculated lattice parameters of LaFe13-xTx (T = Al, Si) compounds are in good agreement with the ex- perimental data. Qualitative analyses are carried out on the behavior of the Curie temperature and magnetocrystalline anisotropy. All the results indicate that the pair potentials based on the lattice inversion method can effectively give a deeper insight into the structure and property of complex materials. Keywords: Rare Earth Compounds, Interatomic Potentials, Site Preference, Computers Simulation 1. Introduction Recently rare earth transition metal intermetallic com- pounds with NaZn13-type structure have attracted con- siderable attention owing to their potential application and intriguing magnetic properties [1-4]. In rare earth- transition metal intermetallic compounds the strong magnetic coupling between 3d atoms gives rise to the large magnetization. It seems that the novel R-T inter- metallic compounds with excellent magnetic properties are always related to a high content of 3d transition metal. Among all binary R-M intermetallic compounds only LaCo13 crystallizes in the cubic NaZn13-type structure (space group Fm-3c). The LaFe13 compound does not exist. Generally, the positive heat of alloying between La and Fe is considered to be the reason why no LaFe13 bi- nary compound exists. Up to now, many compounds with NaZn13-type structure have been obtained in R (T, M) 13 systems (R = rare earth, T = Fe, Co, Ni and M = Al, Si) by elemental substitution. By now ternary compounds LaFe13-xSix (1.3 < x < 2.6) [5] and LaFe13-xAlx (1 < x < 7) [6] are known. Besides, recently the NdFe13-xBex com- pounds (2< x <4) have been synthesized. The cubic symmetry of the structure usually implies a lack of significant magnetocrystalline anisotropy, so that intermetallic compounds with the NaZn13-type structure cannot be used for permanent magnets. Much larger magnetocrystalline anisotropy is expected in materials with a strong anisotropic crystal structure. It has been observed that in some range of Si or Al concentrations the crystal symmetry of the R(M,T)13 compounds trans- forms from cubic (NaZn13-type) to tetragonal in the LaCo13-xSix and PrFe13-xSix compounds (Ce2Ni17Si9-type) [7] or to orthorhombic in the LaFe13-xAlx compounds[8] and hence creates an opportunity for developing magne- tocrystalline anisotropy in 1:13 alloys. In this work, a series of inter-atomic pair potentials in LaFe13-xTx were determined using a general lattice-inversi- on technique with a first principles-based crystal cohe- sive energy calculation. In this way, the stability of LaFe13-xTx and the site preference were evaluated and analyzed. Section 2 given an introduction to the method- ology for the calculation. Section 3 shows the calculated results and give a comparison with the results of the ex- periments. The conclusions and the further discussion are given in the Section 4. 2. Calculation Methods In general, any interatomic pair potential can be obtained by a strict lattice inversion of cohesive curve, and the cohesive energy curves can be obtained either by first  Theoretical Study of the Rare Earth Compounds LaFeT(T= Cr, Cu, Ga, Mn, Ni) and Curie Temperature Variation1167 13-x x principle calculation or by experimental data fitting. The lattice inversion theorem is focused on explaining how to use Chen’s lattice inversion theorem to obtain the intera- tomic pair potential from the first principle cohesive en- ergy curve. A brief introduction of the lattice inversion theorem is proposed as follows [9]: Suppose that the crystal cohesive energy obtained by the first principle calculation is expressed as: E(x) = 00 1 1[()] 2n rbnx (1) where x is the near-neighbor distance, r0(n) is the n neighbor coordination number, b0(n)x is the distance be- tween the central reference atom and its nth neighbor, and [b0(n)x] is the pair potential. A multiplicative closed semi-group b(n). In the process, a lot of virtual lattice points are involved, but the corresponding virtual coordination number is zero. In the b(n), for any two integers m and n, there is a sole integer k such that b(k) = b(m) b(n). Hence, equation (1) can be rewritten as E(x) = 0 1 1[ ()] 2n rbnx (2) where 1 0() () 0 rbbn n rn (3) 0 0 () () () () bnb n bnb n Then the general equation for the interatomic pair po- tential obtained from the inversion can be expressed as () 2()[()] ni InEbnx (4) where I(n) has the characteristics of 1 1 ()/() () () () k bn bk bk Inr bbn (5) I(n) is uniquely determined by a geometrical crystal structure, not related to the concrete element category. Thus, the interatomic pair potentials can be obtained from the known cohesive energy function E(x).The in- teratomic pair potential in distinct atoms can be obtained by the same inversion method, and they are used to study the rare earth intermetallics structures. Which are close to the Morse function, that is: 0 12 0 x= 2 xx RR ФDe e 0 1 (6) where D0 is the depth of potential of potential, R0, γ are parameters. Some important potential are show in the Table 1. 3. Calculation Results The structure of LaFe13-xTx (T = Ga, Ni, Mn, Cr, Cu) compounds are simulated by the energy minimization process and realized by the conjugate gradient method with 14 Å as the cut-off radius of the potentials. The re- sults are taken by the arithmetic average for 30 stochastic configurations. To avoid statistic fluctuation, the model is taken as a 2 × 2 × 2 super cell. 3.1. Phase Stability and Lattice Constants of LaFe13-xTx (T = Cr, Cu, Ga, Mn, Ni) Although the pure LaFe13 compound is unstable, it can be considered as the eigentructure of LaFe13-xTx and their interstitial compounds. In the calculation procedure, the initial lattice constants of LaFe13 are randomly chosen in a certain range. Under the control of the interatomic pair potentials, the energy minimization is carried out by us- ing the conjugate gradient method. The results show that the space group maintains Fm-3c or I4/m·cm with NaZn13-type structure in the tolerance range of 0.001 - 0.1 Å. In the structure of LaFe13, we first substitute T (T = Cr, Cu, Ga, Mn, Ni) atom for Fe in each site with different concentrations and then use the energy minimization method to relax the systems under the interaction of the potentials. With different fractions of the third element, LaFe13-xTx may crystallize in the cubic structure with the space group Fm-3c or in the tetragonal structure with the space group I4/mcm. Thus the average cohesive energy of final structure can be investigated and compared. The average cohesive energies of different structures are displayed in Figure 1. It can be seen that the crystal energy gradually decrease with increasing content of the ternary element T (T = Cr, Cu, Ga, Mn, Ni). The struc- ture stability is judged by the energy and tolerance. The tolerance, which represents the displacement of the atomic position in order to retrieve the space group, is an assistant criterion. Numerous calculations show that, when the tolerance is much large than 0.5 Å,the com- pounds do not exist in experiments. In fact, the site oc- cupation is determined by the energy and the too large tolerance means that the final stable structure has devi- ated too far from the selected one and that the structure has been changed. In the calculation, for LaFe13-xCrx, Cr entering into 96i site of the cubic structure has the lowest energy and the tolerance is acceptable for 0 ≤ x ≤ 3.5. On the other hand, in the range 0 ≤ x ≤ 3.5, the tolerance for Cr entering into 16k site of the tetragonal structure is so large that the tetragonal structure could not exist. However, when 3.5 ≤ x ≤ 4.0, the tolerance with Cr entering into 16 k site of the tetragonal structure drops sharply and the tetragonal Copyright © 2011 SciRes. MSA 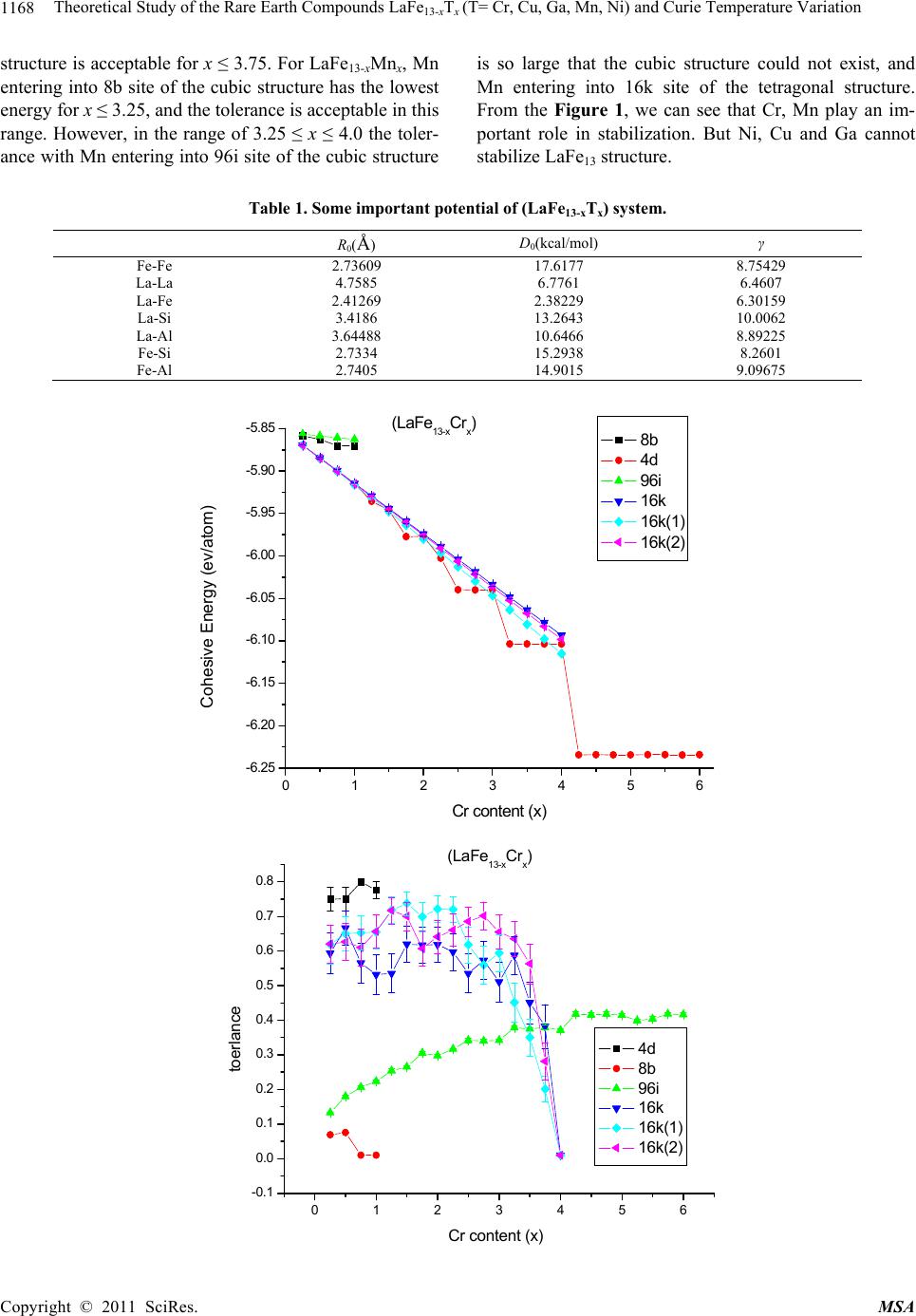 Theoretical Study of the Rare Earth Compounds LaFe13-xTx (T= Cr, Cu, Ga, Mn, Ni) and Curie Temperature Variation Copyright © 2011 SciRes. MSA 1168 structure is acceptable for x ≤ 3.75. For LaFe13-xMnx, Mn entering into 8b site of the cubic structure has the lowest energy for x ≤ 3.25, and the tolerance is acceptable in this range. However, in the range of 3.25 ≤ x ≤ 4.0 the toler- ance with Mn entering into 96i site of the cubic structure is so large that the cubic structure could not exist, and Mn entering into 16k site of the tetragonal structure. From the Figure 1, we can see that Cr, Mn play an im- portant role in stabilization. But Ni, Cu and Ga cannot stabilize LaFe13 structure. Table 1. Some important potential of (LaFe13-xTx) system. R0(Å) D0(kcal/mol) γ Fe-Fe 2.73609 17.6177 8.75429 La-La 4.7585 6.7761 6.4607 La-Fe 2.41269 2.38229 6.30159 La-Si 3.4186 13.2643 10.0062 La-Al 3.64488 10.6466 8.89225 Fe-Si 2.7334 15.2938 8.2601 Fe-Al 2.7405 14.9015 9.09675 0123456 -6.25 -6.20 -6.15 -6.10 -6.05 -6.00 -5.95 -5.90 -5.85 (LaFe13-xCrx) Cohesive Energy (ev/atom) Cr content (x) 8b 4d 96i 16k 16k(1) 16k(2) 0123456 -0.1 0.0 0.1 0.2 0.3 0.4 0.5 0.6 0.7 0.8 (LaFe13-xCrx) toerlance Cr content (x) 4d 8b 96i 16k 16k(1) 16k(2) 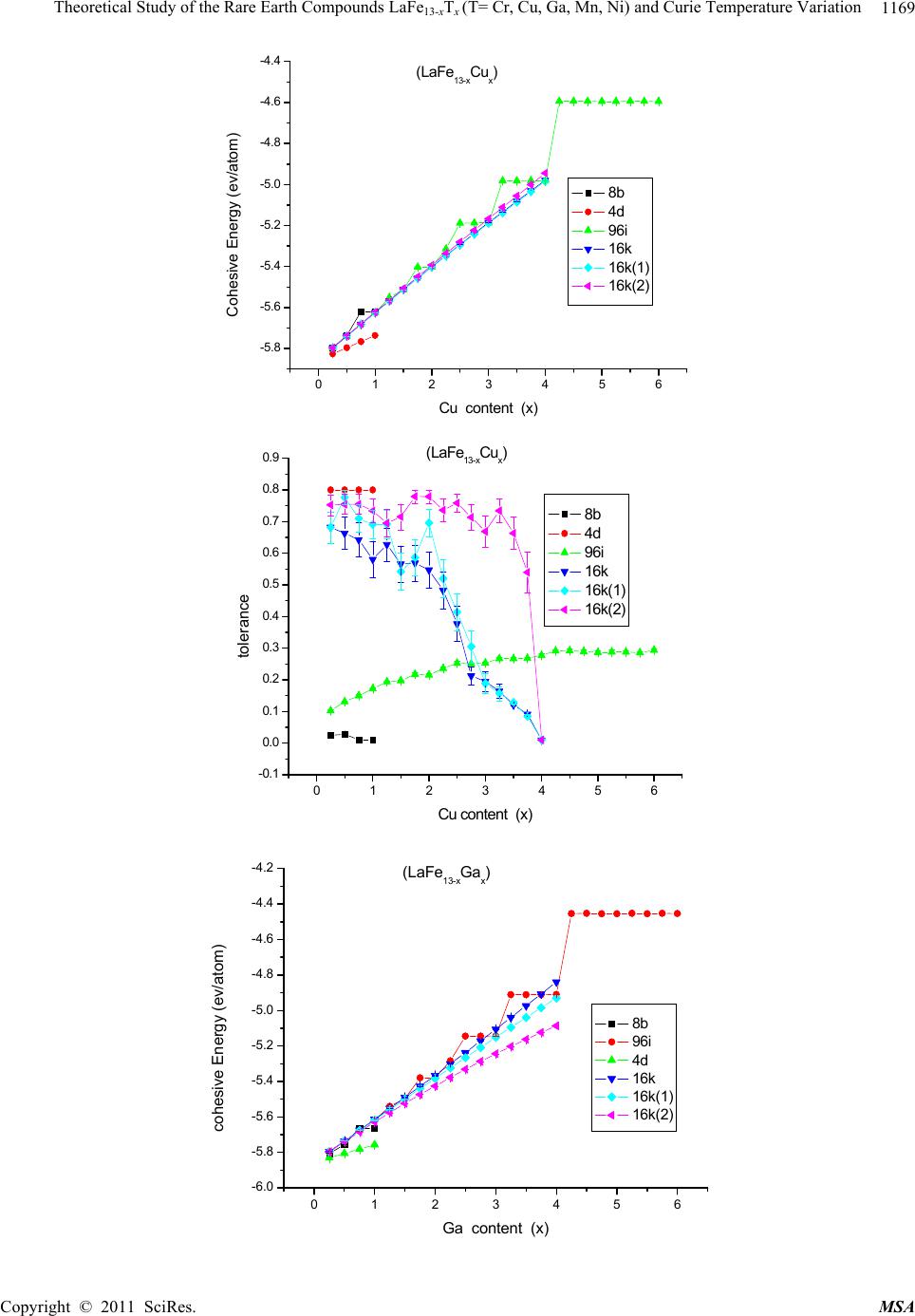 Theoretical Study of the Rare Earth Compounds LaFeT(T= Cr, Cu, Ga, Mn, Ni) and Curie Temperature Variation1169 13-x x 0123456 -5.8 -5.6 -5.4 -5.2 -5.0 -4.8 -4.6 -4.4 (LaFe13-xCux) Cohesive Energy (ev/atom) Cu content (x) 8b 4d 96i 16k 16k(1) 16k(2) 0123456 -0.1 0.0 0.1 0.2 0.3 0.4 0.5 0.6 0.7 0.8 0.9 (LaFe13-xCu x) tolerance Cu content (x) 8b 4d 96i 16k 16k(1) 16k(2) 0123456 -6.0 -5.8 -5.6 -5.4 -5.2 -5.0 -4.8 -4.6 -4.4 -4.2 (LaFe13-xGax) cohesive Energy (ev/atom) Ga content (x) 8b 96i 4d 16k 16k(1) 16k(2) Copyright © 2011 SciRes. MSA 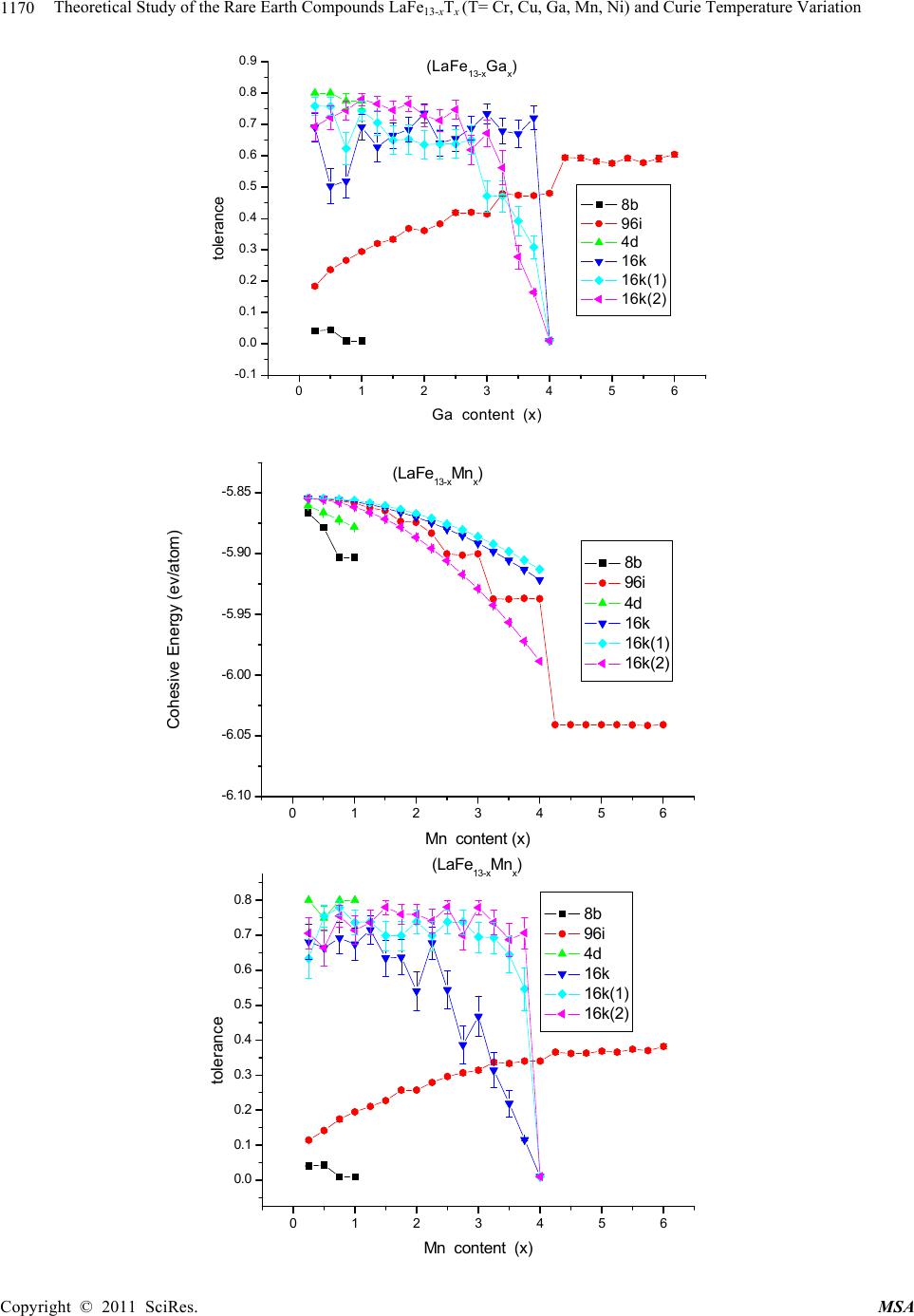 Theoretical Study of the Rare Earth Compounds LaFeT(T= Cr, Cu, Ga, Mn, Ni) and Curie Temperature Variation 1170 13-x x 0123456 -0.1 0.0 0.1 0.2 0.3 0.4 0.5 0.6 0.7 0.8 0.9 (LaFe13-xGax) tolerance Ga content (x) 8b 96i 4d 16k 16k(1) 16k(2) 0123456 -6.10 -6.05 -6.00 -5.95 -5.90 -5.85 (LaFe13-xMn x) Cohesive E ne r gy ( ev /atom ) Mn content (x) 8b 96i 4d 16k 16k(1) 16k(2) 0123456 0.0 0.1 0.2 0.3 0.4 0.5 0.6 0.7 0.8 (LaFe13-xMnx) tolerance Mn c ont ent (x) 8b 96i 4d 16k 16k(1) 16k(2) Copyright © 2011 SciRes. MSA 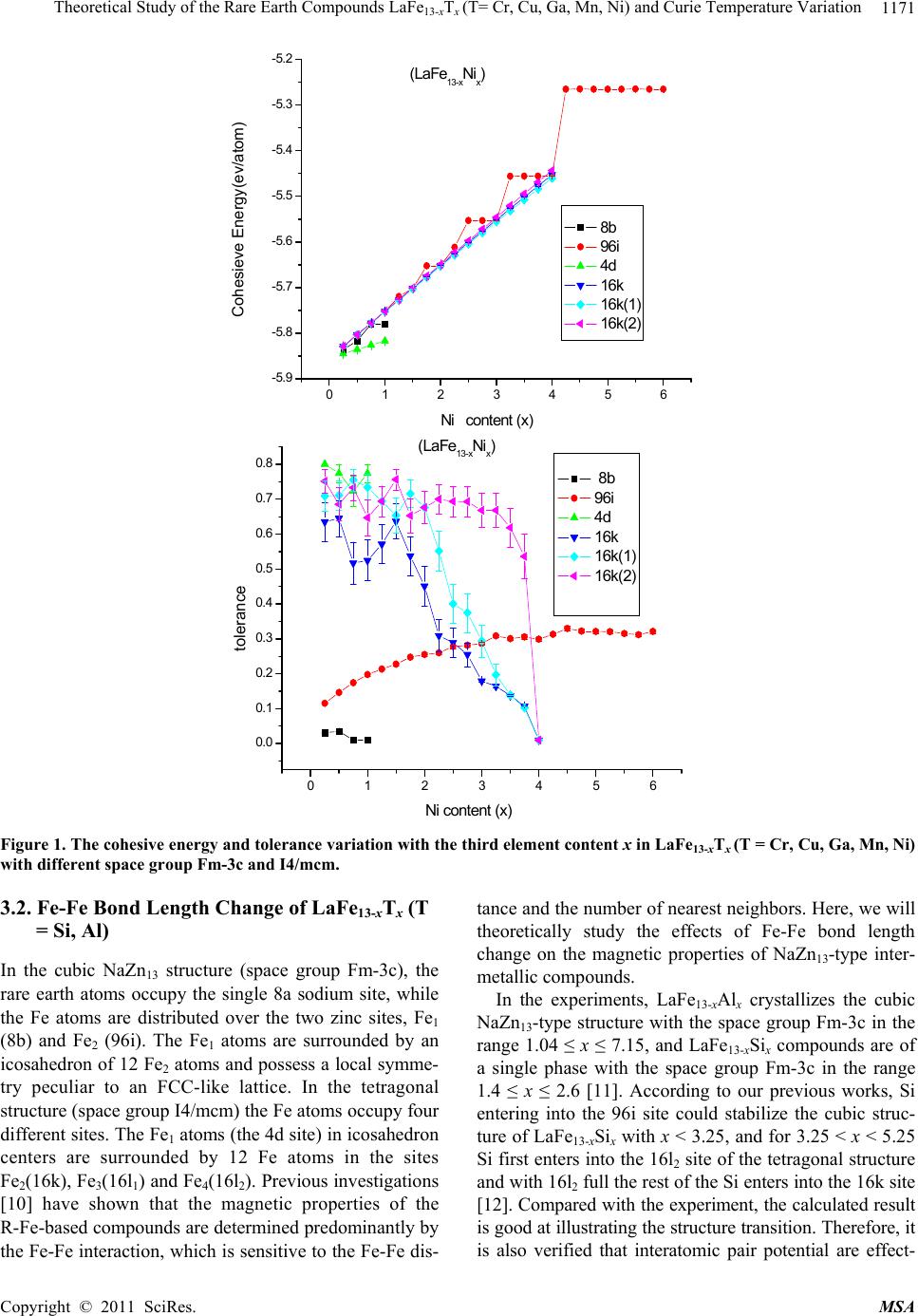 Theoretical Study of the Rare Earth Compounds LaFe13-xTx (T= Cr, Cu, Ga, Mn, Ni) and Curie Temperature Variation Copyright © 2011 SciRes. MSA 1171 0123456 -5.9 -5.8 -5.7 -5.6 -5.5 -5.4 -5.3 -5.2 (LaFe13-xNix) Cohesieve Energy(ev/atom) N i con te nt (x) 8b 96i 4d 16k 16k(1) 16k(2) 0123456 0.0 0.1 0.2 0.3 0.4 0.5 0.6 0.7 0.8 (LaFe13-xNix) tolerance Ni content (x) 8b 96i 4d 16k 16k(1) 16k(2) Figure 1. The cohesive energy and tolerance variation with the third element content x in LaFe13-xTx (T = Cr, Cu, Ga, Mn, Ni) with different space group Fm-3c and I4/mcm. 3.2. Fe-Fe Bond Length Change of LaFe13-xTx (T = Si, Al) tance and the number of nearest neighbors. Here, we will theoretically study the effects of Fe-Fe bond length change on the magnetic properties of NaZn13-type inter- metallic compounds. In the cubic NaZn13 structure (space group Fm-3c), the rare earth atoms occupy the single 8a sodium site, while the Fe atoms are distributed over the two zinc sites, Fe1 (8b) and Fe2 (96i). The Fe1 atoms are surrounded by an icosahedron of 12 Fe2 atoms and possess a local symme- try peculiar to an FCC-like lattice. In the tetragonal structure (space group I4/mcm) the Fe atoms occupy four different sites. The Fe1 atoms (the 4d site) in icosahedron centers are surrounded by 12 Fe atoms in the sites Fe2(16k), Fe3(16l1) and Fe4(16l2). Previous investigations [10] have shown that the magnetic properties of the R-Fe-based compounds are determined predominantly by the Fe-Fe interaction, which is sensitive to the Fe-Fe dis- In the experiments, LaFe13-xAlx crystallizes the cubic NaZn13-type structure with the space group Fm-3c in the range 1.04 ≤ x ≤ 7.15, and LaFe13-xSix compounds are of a single phase with the space group Fm-3c in the range 1.4 ≤ x ≤ 2.6 [11]. According to our previous works, Si entering into the 96i site could stabilize the cubic struc- ture of LaFe13-xSix with x < 3.25, and for 3.25 < x < 5.25 Si first enters into the 16l2 site of the tetragonal structure and with 16l2 full the rest of the Si enters into the 16k site [12]. Compared with the experiment, the calculated result is good at illustrating the structure transition. Therefore, it is also verified that interatomic pair potential are effect-  Theoretical Study of the Rare Earth Compounds LaFeT(T= Cr, Cu, Ga, Mn, Ni) and Curie Temperature Variation 1172 13-x x Table2. The crystallographic parameters a and c of LaFe13-xTx (Al, Si) in Å, obtained from experiments (exp.) and calcula- tions (cal.), and Ref. is the source of the experimental data. x a(cal) c(cal) a(exp) c(exp) Ref. LaFe13-xSix 2 11.470 11.463 [13] 3 8.035 11.485 7.994 11.363 4 7.997 11.501 7.974 11.542 5 7.967 11.595 7.954 11.720 LaFe13-xAlx 2 11.547 11.668 [14] 3 11.569 11.731 4 11.614 11.794 5 11.688 11.857 6 11.794 11.920 7 11.871 11.983 tive. Table 2 shows the lattice parameters of LaFe13-xTx (T = Si, Al) obtained from both the experiments [13] [14] and calculations. The results correspond well to experi- mental data. The bond lengths of Fe-Fe which play an important role in magnetic properties can be evaluated. In this subsection, the dependence of Curie temperature on the Si/Al concentration is analyzed qualitatively. In the experiment, the value of TC for the LaFe11.5Si1.5 compound is 188 K. Just as Givord et al. have pointed out, there are two types of exchange interactions, positive and negative, which depend on the length of Fe-dumb- bells. All the exchange interactions could be positive except the case associated with 96i-96i, 96i-8b, since the distances between these iron atoms are small. In order to study the trend of Curie temperature varia- tion, the distance between special sites and the related exchange interaction should be investigated. Figure 2 presents the bond lengths in LaFe13-xSix, indicating that the lengths of 96i-96i are shorter than 2.46 Å which can be considered as the threshold of switching exchange interaction. The distances between 96i-96i and 96i-8b decrease with increasing Si content, which are similar to that in the experimental trend. Notice that the bond lengths of 96i-8b are larger than 2.46 Å, then the varia- tion of exchange interaction from 96i-8b pairs can be neglected. As far as iron pairs except 96i-96i are con- cerned, the positive exchange interactions associated with large bond lengths are weak, even though the intro- duction of Si atom has the effect of reduction on them. Then the contribution to the exchange interaction from these iron pairs can also be neglected. In the next step, we will focus on the effect of 96i-96i lengths based on the variation of Curie temperature. In the above subsection, the analysis of site preference behavior shows that Si atoms have a pronounced prefer- ence for occupying 96i site. On the one hand, the number of 96i-96i (Fe-Fe) dumbbell pairs reduces and the nega- tive exchange interaction with the increase of Si atom, and in thus beneficial in improving the Curie temperature. on the other hand, bond lengths of 96i-96i pairs decrease with increasing Si content, and thus enhance the negative exchange interaction and lead to a lower Curie tempera- ture. So there is a competing mechanism arising from Si content increase, i.e. the variation of Curie temperature is not monotonic with the increment of Si concentration. Givord et al. [15] have shown that the threshold of switching Fe-Fe exchange interaction is 2.45 Å. In other words, if the distance of Fe-Fe is shorter than 2.45 Å, then the exchange interaction would be negative, if it is larger than 2.45 Å, then it would be positive. In the pre- sent calculation, the threshold of Fe-Fe exchange interac- tion is taken as 2.46 Å, instead of 2.45 Å from the ex- perimental analysis. This difference may be caused by a systemic error in derivation of these quasi-in- teratomic potentials. ab initio 4. Conclusions and Discussions Pair potentials based on calculations and on the lattice-inversion method have been used to calculate the structural properties of a series of LaFe13-xTx (T = Cr, Cu, Ga, Mn, Ni).The results show that Cr and Mn make the cohesive energy decrease markedly, indicating that these atoms can stabilize LaFe13-xTx with the cubic NaZn13-type or its derivative tetragonal structure. ab initio Furthermore, the increase of the Curie temperature of LaFe13-xTx (T = Si, Al) with increasing Si concentration can be qualitatively explained in terms of the distance dependence of the exchange interaction. The presented results indicate that simple pair potentials are useful for studying the structural properties of these kinds of inter- metallics. 5. Acknowledgements The authors would like to thank the stimulating discus- sions with Professor N. X. Chen, and Y. Chen. The pre- sent work is supported by National 973 project Copyright © 2011 SciRes. MSA 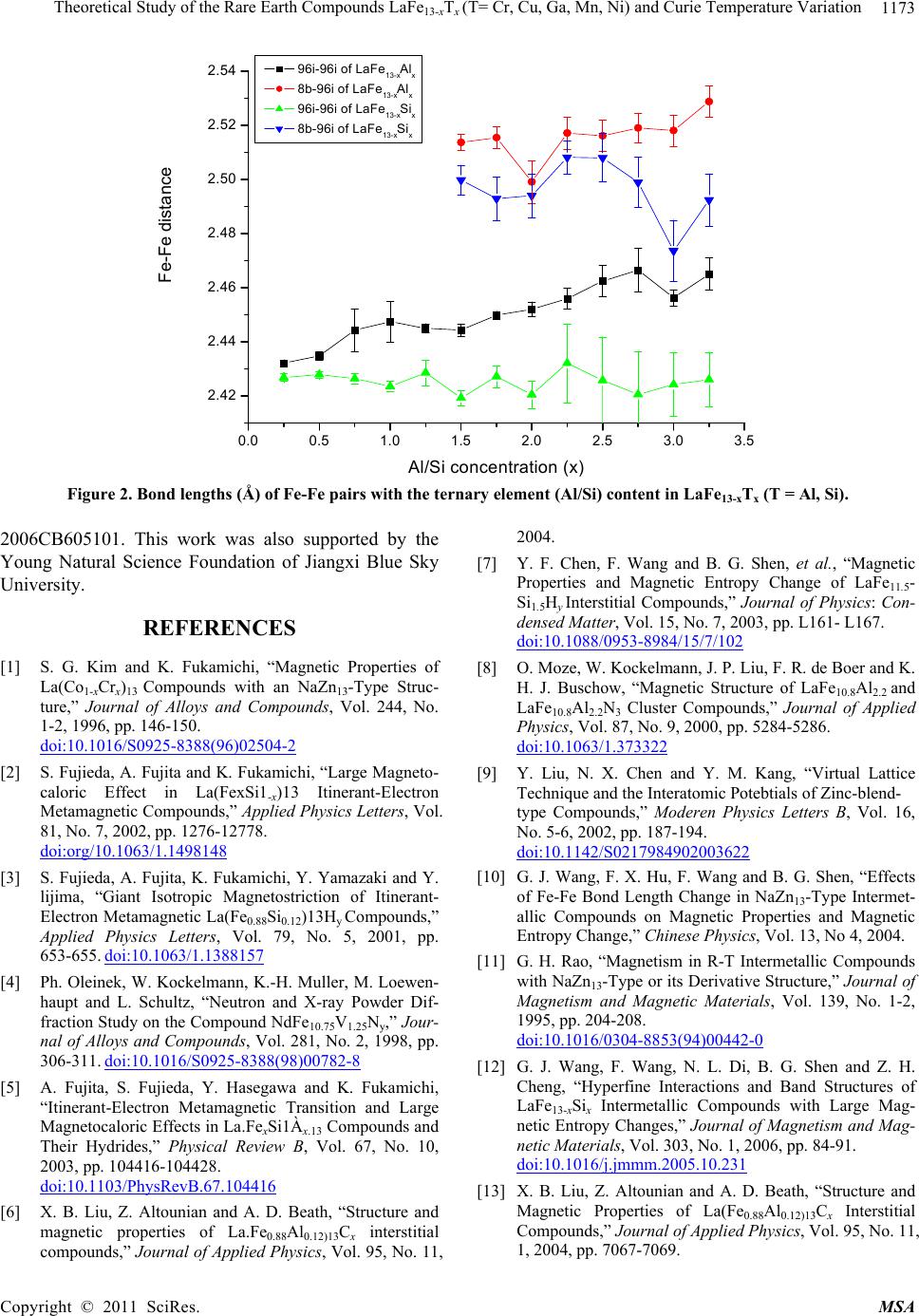 Theoretical Study of the Rare Earth Compounds LaFeT(T= Cr, Cu, Ga, Mn, Ni) and Curie Temperature Variation 1173 13-x x 0.0 0.5 1.0 1.5 2.0 2.5 3.0 3.5 2.42 2.44 2.46 2.48 2.50 2.52 2.54 Fe-Fe distance Al/Si concentration (x) 96i-96i of LaFe13-xAlx 8b-96i of LaFe13-xAlx 96i-96i of LaFe13-xSix 8b-96i of LaFe13-xSix Figure 2. Bond lengths (Å) of Fe-Fe pairs with the ternary element (Al/Si) content in LaFe13-xTx (T = Al, Si). 2006CB605101. This work was also supported by the Young Natural Science Foundation of Jiangxi Blue Sky University. REFERENCES [1] S. G. Kim and K. Fukamichi, “Magnetic Properties of La(Co1-xCrx)13 Compounds with an NaZn13-Type Struc- ture,” Journal of Alloys and Compounds, Vol. 244, No. 1-2, 1996, pp. 146-150. doi:10.1016/S0925-8388(96)02504-2 [2] S. Fujieda, A. Fujita and K. Fukamichi, “Large Magneto- caloric Effect in La(FexSi1-x)13 Itinerant-Electron Metamagnetic Compounds,” Applied Physics Letters, Vol. 81, No. 7, 2002, pp. 1276-12778. doi:org/10.1063/1.1498148 [3] S. Fujieda, A. Fujita, K. Fukamichi, Y. Yamazaki and Y. lijima, “Giant Isotropic Magnetostriction of Itinerant- Electron Metamagnetic La(Fe0.88Si0.12)13Hy Compounds,” Applied Physics Letters, Vol. 79, No. 5, 2001, pp. 653-655. doi:10.1063/1.1388157 [4] Ph. Oleinek, W. Kockelmann, K.-H. Muller, M. Loewen- haupt and L. Schultz, “Neutron and X-ray Powder Dif- fraction Study on the Compound NdFe10.75V1.25Ny,” Jour- nal of Alloys and Compounds, Vol. 281, No. 2, 1998, pp. 306-311. doi:10.1016/S0925-8388(98)00782-8 [5] A. Fujita, S. Fujieda, Y. Hasegawa and K. Fukamichi, “Itinerant-Electron Metamagnetic Transition and Large Magnetocaloric Effects in La.FexSi1Àx.13 Compounds and Their Hydrides,” Physical Review B, Vol. 67, No. 10, 2003, pp. 104416-104428. doi:10.1103/PhysRevB.67.104416 [6] X. B. Liu, Z. Altounian and A. D. Beath, “Structure and magnetic properties of La.Fe0.88Al0.12)13Cx interstitial compounds,” Journal of Applied Physics, Vol. 95, No. 11, 2004. [7] Y. F. Chen, F. Wang and B. G. Shen, et al., “Magnetic Properties and Magnetic Entropy Change of LaFe11.5- Si1.5Hy Interstitial Compounds,” Journal of Physics: Con- densed Matter, Vol. 15, No. 7, 2003, pp. L161- L167. doi:10.1088/0953-8984/15/7/102 [8] O. Moze, W. Kockelmann, J. P. Liu, F. R. de Boer and K. H. J. Buschow, “Magnetic Structure of LaFe10.8Al2.2 and LaFe10.8Al2.2N3 Cluster Compounds,” Journal of Applied Physics, Vol. 87, No. 9, 2000, pp. 5284-5286. doi:10.1063/1.373322 [9] Y. Liu, N. X. Chen and Y. M. Kang, “Virtual Lattice Technique and the Interatomic Potebtials of Zinc-blend- type Compounds,” Moderen Physics Letters B, Vol. 16, No. 5-6, 2002, pp. 187-194. doi:10.1142/S0217984902003622 [10] G. J. Wang, F. X. Hu, F. Wang and B. G. Shen, “Effects of Fe-Fe Bond Length Change in NaZn13-Type Intermet- allic Compounds on Magnetic Properties and Magnetic Entropy Change,” Chinese Physics, Vol. 13, No 4, 2004. [11] G. H. Rao, “Magnetism in R-T Intermetallic Compounds with NaZn13-Type or its Derivative Structure,” Journal of Magnetism and Magnetic Materials, Vol. 139, No. 1-2, 1995, pp. 204-208. doi:10.1016/0304-8853(94)00442-0 [12] G. J. Wang, F. Wang, N. L. Di, B. G. Shen and Z. H. Cheng, “Hyperfine Interactions and Band Structures of LaFe13-xSix Intermetallic Compounds with Large Mag- netic Entropy Changes,” Journal of Magnetism and Mag- netic Materials, Vol. 303, No. 1, 2006, pp. 84-91. doi:10.1016/j.jmmm.2005.10.231 [13] X. B. Liu, Z. Altounian and A. D. Beath, “Structure and Magnetic Properties of La(Fe0.88Al0.12)13Cx Interstitial Compounds,” Journal of Applied Physics, Vol. 95, No. 11, 1, 2004, pp. 7067-7069. Copyright © 2011 SciRes. MSA  Theoretical Study of the Rare Earth Compounds LaFeT(T= Cr, Cu, Ga, Mn, Ni) and Curie Temperature Variation 1174 13-x x [14] H. Chang, N. X. Chen, J. K. Liang and G. H. Rao, “The- oretical Study of Phase Forming of NaZn13-Type Rare- Earth Intermetallics,” Journal of Physics: Conden- sed Matter, Vol. 15, No. 2, 2003, pp. 109-120. doi:10.1088/0953-8984/15/2/311 [15] Y. F. Chen, F. Wang, B. G. Shen, F. X. Hu, J. R. Sun, G. J. Wang and Z. H. Cheng, “Magnetic Properties and Magnetic Entropy Change of LaFe11.5Si1.5Hy Interstitial Compounds,” Journal of Physics: Condensed Matter, Vol. 15, No. 7, 2003, pp. L161-L167. doi:10.1088/0953-8984/15/7/102 Copyright © 2011 SciRes. MSA
|