310 H. Srivastava et al. / J. Biomedical Science and Engineering 2 (2009) 304-311
SciRes Copyright © 2009 JBiSE
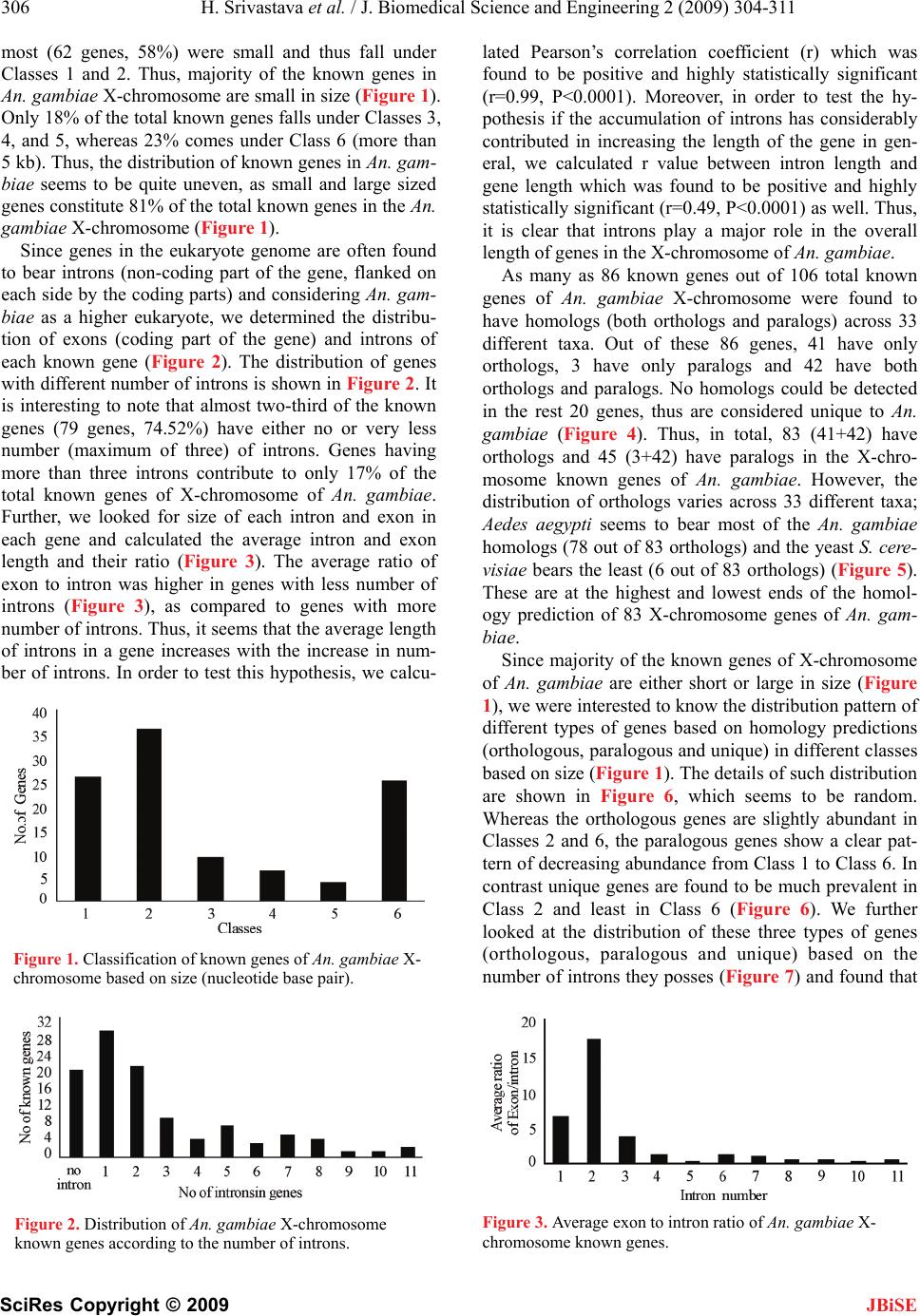
servation of this gene is exactly overlaps with the posi-
tion of taxa in the phylogenetic tree.
In conclusion, the present study not only provides
fine-scale views to the genetic architecture of the X-
chromosome of the model malaria vector of African
importance, but also reveals several interesting features
on evolutionary insights into genes and taxa of different
taxonomic status. The information is of great importance,
especially to the population geneticists, to understand
genetic diversity and infer the respective roles of de-
mography and natural selection in evolution of genes in
different Anopheles species populations of local impor-
tance.
5. ACKNOWLEDGEMENTS
Extramural funding from the Indian Council of Medical Research
(ICMR), New Delhi in the form of an Ad-hoc research grant to AD is
thankfully acknowledged. We thank Dr. Neena Valecha for her kind
support to HS during the initial phase of the study. Lily Basu, Deep-
shikha Lal, Garima Goyal and Suchita Singh helped in organizing the
initial work elements.
REFERENCES
[1] Hahn, M. W., Han, M. V., and Han, S. G., (2007) Gene
family evolution across 12 Drosophila genome, Public
Library of Science Genetics, 3, 1–12.
[2] Matthee, C. A., Eick, G., Willows, M. S., Montgelard, C.,
Pardini, A. T., and Robinson, T. J., (2007) Indel evolu-
tion of mammalian introns and the utility of non coding
nuclear markers in eutherian phylogenetics, Molecular
Phylogenetics and Evolution, 42, 827–837.
[3] Cardazzo, B., Bargelloni, L., Toffolatti, L., and Patar-
nello, T., (2003) Intervening sequences in paralogous
genes: A comparative genomic approach to study the
evolution of X chromosome introns, Molecular Biology
and Evolution, 20, 2034–2041.
[4] Gazave, E., Bonet, T. M., Fernando, O., Charlesworth, B.,
and Navarro, A., (2007) Patterns and rates of intron di-
vergence between humans and chimpanzees, Genome
Biology, 8, 1–13.
[5] Huynen, M. A. and Bork, P., (1998) Measuring genome
evolution, Proceedings of National Academy of Sciences
USA, 95, 5849–5856.
[6] Clark, A. G., Eisen, B. M., Smith, D. R., Bergman, C. M.,
Oliver, B., Markow, T. A., et al., (2007) Evolution of
genes and genomes on the Drosophila phylogeny, Nature,
450, 203–218.
[7] WHO. (2005) World malaria report.
http://www.rbm.who.int/wmr.
[8] Zakeri, S., Afsharpad, M., Raeisi, A., and Djadid, N. D.,
(2007) Prevalence of mutations associated with antima-
larial drugs in Plasmodium falciparum isolates prior to
the introduction of sulphadoxine-pyrimethamine as first-
line treatment in Iran, Malaria Journal, 6, 1–2.
[9] Holt, R. A., Subramanian, G. M., Helpern, A., Sutton, G.
G., Charlab, R., Nusskern, D. R., et al., (2002) The ge-
nome sequence of the malaria mosquito Anopheles gam-
biae, Science, 298, 129–149.
[10] Stephen, S. F., (2004) The X chromosome in population
genetics, Nature Reviews Genetics, 5, 43–51.
[11] Vogl, C., Das, A., Beaumont, M., Mohanty, S., and
Stephan, W., (2003) Population subdivision and molecu-
lar sequence variation: Theory and and analysis of Dro-
sophila ananassae data, Genetics, 165, 1385–1395.
[12] Bains, J. F., Das, A., Mousset, S., and Stephan, W.,
(2004) The role of natural selection in genetic differen-
tiation of worldwide populations of Drosophila ananas-
sae, Genetics, 168, 1987–1998.
[13] Das, A., Mohanty, S., and Stephan, W., (2004) Inferring
the population structure and demography of Drosophila
ananassae from multilocus data, Genetics, 168, 1975–
1985.
[14] Castillo-Davis, C. I., Mekhedov, S. L., Hartl, D. L.,
Koonin, E. V., and Kondrashov, F. A., (2002) Selection
for short introns in highly expressed genes, Nature Ge-
netics, 31, 414–418.
[15] Vinogrado, A. E. (2004) Compactness of human house-
keeping genes: Selection for economy or genomic design,
Trends in Genetics, 20, 248–253.
[16] Jain, M., Tyagi, A. K., and Khurana, J. P., (2006) Ge-
nome wide analysis, evolutionary expansion, and expres-
sion of early auxin-responsive SAUR gene family in rice
(Oryza sativa), Genomics, 88, 360–371.
[17] Jain, M., Khurana, P., Tyagi, A. K., and Khurana, J.,
(2007) Genome-wide analysis of intronless genes in rice
and Arabidopsis, Functional & Integrative Genomics, In
Press.
[18] Simpson, A. G. B., Macquarrie, E. K., Roger, A. J.,
(2002) Eukaryotic evolution: Early origin of canonical
introns, Nature, 419, 270.
[19] Jones, A. K., Grauso, M., and Sattelle, B. D., (2004) The
nicotinic acetylcholine receptor gene family of the ma-
laria mosquito, Anopheles gambiae, Genomics, 85, 176–
187.
[20] Matsuda, K., Buchigham, S. D., Kleier, D., Rauh, J. J.,
Grauso, M., and Sattelle, D. B., (2001) Neonicotinoids:
Insecticides acting on insect nicotinic acetylcholine re-
ceptors, Trends in Pharmacological Sciences, 22,
573–580.
[21] Bond, G. J., Marina, C. F., and Williams, T., (2004) The
naturally derived insecticide spinosad is highly toxic to
Aedes and Anopheles mosquito larvae, Medical and
Veterinary Entomology, 18, 50–56.
[22] Manuel, I., David, P., and Scott, W. R., (2007) Coevolu-
tion of genomic intron number and splice sites, Trends in
Genetics, 23, 321–325.
[23] Jordan, I. K., Marino-Ramirez, L., Wolf, Y. I., and
Koonin, E. V., (2004) Conservation and co-evolution in
the scale-free human gene co-expression network, Mole-
culer Biologyand Evolution, 21, 2058–2070.
[24] Carmel L., Rogozin I. B., Wolf, Y. I., and Koonin, E. V.,
(2007) Evolutionarily conserved genes preferentially ac-
cumulate introns, Genome Research, 17, 1045–1050.
[25] Castillo-Davis, C. I., Bedford, T. B. C., and Hartl, D. L.,
(2004) Accelerated rates of intron gain/loss and protein
evolution in duplicate genes in human and mouse malaria
parasite, Molecular Biology and Evolution, 21, 1422–
1427.
[26] Stirling, B., Yang, Z. K., Gunter, L. E., Tuskan, G. A.,
and Bradshaw, H. D., (2003) Comparative sequence