Paper Menu >>
Journal Menu >>
 J. Biomedical Science and Engineering, 2008, 1, 141-146 Published Online August 2008 in SciRes. http://www.srpublishing.org/journal/jbise JBiSE Prediction of human microRNA hairpins using only positive sample learning Dang Hung Tran*, 1, Tho Hoan Pham2, Kenji Satou1, 3 & Tu Bao Ho1 1Japan Advanced Institute of Science and Technology, 1-1 Asahidai, Nomi, Ishikawa 923-1292 J apan. 2Han oi National Unive rs i ty of Education, 136 Xuan Thuy, Hanoi, Viet Nam. 3Kanazawa University, Kakuma, Kanazawa 920-1192, Japan. *Corresponden ce should be addressed to Dang Hung T r an (hungtd@jaist.ac.jp). ABSTRACT MicroRNAs (miRNAs) are small molecular non-coding RNAs that have important roles in the post-transcriptional mechanism of animals and plants. They are commonly 21-25 nucleo- tides (nt) long and derived from 60-90 nt RNA hairpin structures, called miRNA hairpins. A lar- ger number of sequence segments in the human genome have been computationally identified with such 60-90 nt hairpins, however the major- ity of them are not miRNA hairpins. Most exist- ing computational methods for predicting miRNA hairpins are based on a two-class classi- fier to distinguish between miRNA hairpins and other sequence segments with hairpin struc- tures. The difficulty of these methods is how to select hairpins as negative examples of miRNA hairpins in the training dataset, since only a few miRNA hairpins are available. Therefore, these classifiers may be mis-trained due to some false negative examples of the training dataset. In this paper, we introduce a one-class support vector machine (SVM) method to predict miRNA hair- pins among the hairpin structures. Different from existing methods for predicting miRNA hairpins, the one-class SVM classifier is trained only on the information of the miRNA class. We also il- lustrate some examples of predicting miRNA hairpins in human chromosomes 10, 15, and 21, where our method overcomes the above disad- vantages of existing two-class methods. Keywords: MicroRNA; Hairpin; One-class SVM 1. INTRODUCTION MicroRNAs (miRNAs) are small, non-coding RNAs (21- 25 nucleotides in length) that regulate the expression of protein-encoding genes at the post-transcriptional level [1, 2, 21]. Each miRNA derives from a larger precursor, which folds into an imperfect stem-loop structure. In human, the processing and maturation of miRNAs are divided into several steps before silencing their targets. First, the long primary transcripts (pri-miRNAs), which can be up to several kilobases, are processed by Drosha- complex in nucleus to yield precursor miRNAs (pre- miRNAs) [10, 12]. The pre-miRNA is a double-stranded sequence of about 60-90 nt with a 2-nt 3' overhang and forms a hairpin structure (also called miRNA hairpin). Second, pre-miRNAs are transported from the nucleus into the cytoplasm by another complex, which consists of Exportin 5 and RanGTP [6, 29]. Subsequently, the pre- miRNA is cleaved into an imperfect double-stranded RNA duplex by endonuclease RNase III enzyme called Dicer [25, 29, 42]. This duplex is composed of the mature miRNA strand and its complementary strand. Finally, mature miRNAs are incorporated into RICS (RNA- induced silencing complex) before they bind to their tar- gets to regulate gene expression. Until now, several computational approaches have been proposed for predicting miRNAs. Most of them are based on the common structural characteristic of secon- dary structures of their pre-miRNAs [15, 35, 40]. Since pre-miRNAs are often short (60-90 nt), there can be too many subsequences in a genome having hairpin structures. However, only a minority of them are miRNA hairpins. Using only information of their structures therefore may not allow us to distinguish miRNA hairpins from other hairpin structures. Other methods that consider informa- tion of both sequences and structures are needed. Most methods so far used a two-class classifier to sepa- rate the miRNA hairpins from the ones assumed to be negative. The main difference between these methods is how negative examples are selected for the two-class classifier training dataset. For example, Szafranski et al. [35] and Xue et al. [40] selected examples that overlap with one of the last exon of known mRNAs; or Helvik et al. [15] tried to get them randomly from DNA sequences with hairpin structures as ``negative'' miRNA hairpins. The negative examples collected in such ways would con- tain false negatives, since no study so far has mentioned the information regarding true negative miRNA hairpins. In other words, only the information of miRNA hairpins is available. Therefore, the classifier of existing methods may be incorrect, due to some false negative miRNA hairpins contained in the training dataset. In this paper, we present a new method for predicting miRNA hairpins that employs support vector machines SciRes Copyright © 2008  142 D. H. Tran et al. / J. Biomedical Science and Engineering 1 (2008) 141-146 SciRes Copyright © 2008 JBiSE for one-class classification (one-class SVMs). One-class SVMs recently have been successfully applied in several areas, especially domains with imbalanced data such as document classification [30], gene prediction [19] and image retrieval [9]. Different from previous methods for predicting miRNA hairpins, our method uses only avail- able miRNA hairpins for training the model, while other methods train their classifiers by using an additional data- set of negative examples, which may contain some false negatives as explained above. Moreover, more features of hairpin sequences and structures are used to represent hairpins, with expectation that they would be useful for the model. Our one-class SVM classifier gave good re- sults in predicting miRNA hairpins. We also illustrated some examples of predicting miRNA hairpins in human chromosomes 10, 15, and 21 where our method can avoid the problem of false negative examples of the existing two-class methods. 2. MATERIALS AND METHODS 2.1. Datasets for training and testing As mentioned in Section 1, our method uses one-class SVMs to recognize miRNA hairpins from potential ones produced by ScorePin [15]. To do this, the one-class SVM model should capture the characteristics of known miRNA hairpins. In our work, the positive class we used consists of 474 known human miRNA hairpins from miRBase (version 8.1) [13, 14]. (http://microrna. sanger.ac.uk/sequences/) that have been verified by ex- periments or predicted by computational methods with high confidence. To ensure that all miRNA hairpins were folded as hairpins, we removed a few of those containing none or more than one RNAfold-predicted hairpin-loop. The positive class used in this work is therefore of 451 miRNA hairpins. To evaluate our one-class SVM models for miRNA hairpins, we conducted two kinds of experiments. Cross-validation : like some previous researches [15, 35, 40], we first prepared the dataset for the cross-validation procedure to compare our method with the other methods. The dataset contained 451 positive examples as described above, and 727 negative examples of miRNAs hairpins. These 727 negative miRNA hairpins were ScorePin- hairpins that overlap with the last exon of known coding- protein genes. We randomly partitioned the dataset into three subsets, such that the numbers of both positive and negative examples in each of the three subsets were equivalent or nearly equivalent. Of them, one subset was retrained as the validation data for test prediction methods, and was trained on the two remaining subsets (note that with our method, one-class SVM, only positive examples are used for the training). The cross-validation procedure was repeated three times. Results from three trials were then averaged to produce a single estimation. Test on chromosomes 10, 15, and 21: we use all known miRNA hairpins, excluding ones on chromosomes 10, 15 and 21, to train the one-class SVM model. This model is then used to recognize miRNA hairpins from ScorePin- hairpins (see Section 3.1). Table 1 presents a summary of all data sets used in two kinds of experiments. Table 1. The data of human miRNA hairpins. Experiment #Examples Testing on chromosomes 437 training examples 41039 hairpin candidates Cross-validation 2/3 x 451 training positives 1/3 x 451 testing positives 1/3 x 727 testing negatives 2.2. One-class support vector machines Support vector machine (SVM) is a learning technique based on statistical learning theory [38]. It has been ap- plied to a wide range of real-world tasks. The formulation of SVMs can be considered as a simple linear classifica- tion, normally using both negative and positive examples for training. SVMs can perform nonlinear separation by using a kernel technique, which realizes a nonlinear map- ping to a feature space. Scholkopf et al. [33] have ex- tended standard SVMs to one-class classification prob- lems. Their approach is to construct a hyperplane that is maximally distant from the origin [33]. In this section, we give details of the algorithm for training one-class SVMs proposed by Scholkopf et al. [33]. The training algorithm is as follows: let the training data N lRxxx ∈,...,, 21 belong to one class, where i xis a feature vector and l is the number of examples. The one- class SVM estimates a function that will take the valu e +1 in a region where the majority of the data points are con- centrated, and the value -1 everywhere else [30, 33]. For- mally, the function can be written as follows: ⎪ ⎩ ⎪ ⎨ ⎧ ∈− ∈+ =Sxif Sxif xf 1 1 )( where S is a simple subset of input space and S is the complement of.SLet HX → Φ : be a kernel map which converts the training examples from the origin space to a feature space. The strategy is to map the data into the fea- ture space corresponding to the kernel, and to separate them from the origin by the maximum margin. In order to separate the data set from the origin, we need to solve the following quadratic programming problem [9, 30, 33]: ⎪ ⎪ ⎩ ⎪ ⎪ ⎨ ⎧ ≥=−≥Φ −+ ∑ = .0;,...,1,))(.( 1 2 1 min 1 2 iii l i i lixw vl w ξξρ ρξ where )1,0( ∈ v is a parameter that represents an upper bound on the fraction of outliers in the data, ρ is the mar- gin of the hyperplane with respect to the data, and i x are non-zero slack variables allowing a soft margin. We ob- tain w and ρ by solving this problem. When we give a new data point x to be classified, a label is assigned ac-  D. H. Tran et al. / J. Biomedical Science and Engineering 1 (2008) 141-146 143 SciRes Copyright © 2008 JBiSE cording to the decision function, which can be expressed as: )))(.sgn(()( ρ − Φ =i xwxf Instead of solving the primal optimization problem di- rectly, one can consider the following dual program: ⎪ ⎪ ⎩ ⎪ ⎪ ⎨ ⎧ =≤≤ ∑ ∑ .1, 1 0 ),( 2 1 max iii i jiji vl xxK αα αα here, )),((),( jiji xxxxKΦ= are kernels, which allow many more general decision functions when the data are not linearly separable, and the hyperplane can be repre- sented in a feature space. The parameters i α are La- grange multipliers. In our research, we used the LIBSVM (version 2.84) with three types of kernel functions (linear, polynomial, and radial basis (RBF)). This library is an integrated tool for support vector classification and regression which can handle one-class SVM using the algorithm proposed by Scholkopf et al. [33]. The LIBSVM is available at [43]. 2.3. Structural and sequential features of miRNA hairpins There are many miRNA prediction methods which used structural features as key features. However, recent re- ports have shown that the sequence features are important in predicting miRNA hairpins [39, 40]. Xue et al. [40] indicated that the short contiguous subsequences of miRNA hairpin sequences are significantly distinct from other RNA hairpin sequences. For this reason, we pro- pose a set of features that uses both the sequential fea- tures and structural features to characterize the RNA hairpin structure sequences. For sequential features, we extracted features from RNA hairpin sequences using a 5-nucleotide sliding win- dow along an RNA hairpin sequence, and computed the number of occurrences of each 5-gram. As a result, each sequence is represented by a 1,024-dimensional vector of the number of occurrences of all possible 5-grams. In addition, several other features based on the sequences are considered, such as the number of occurrences of each nucleotide (A, C, G, U) in the 5' and 3' arms and GC- content defined as in [35]. For structural features, we extracted them from the sec- ondary structure of each hairpin. The secondary structures are predicted using RNAfold [16]. The structural features used in our method, were introduced in other previous miRNA prediction methods [15, 35]. The features consist of: 1. miRNA hairpin length as the number of nucleotides. 2. Loop size as the number of unpaired bases in the hairpin loop of the predicted secondary structure. 3. Minimum free energy (MFE) as the total free energy of hairpin structure predicted by using RNAfold tool. 4. Paired bases as the number of nucleotides predicted to be in a hydrogen-bonded state. 5. The numbers of nucleotides from 5' site to the loop start. 6. The number of 2-nt overhangs from 5' site and 3' site to loop start. In total, the feature vector, which is input to our one- class SVMs, consists of 1,036 variables. It captures the characteristics of both the sequence and the structure of the RNA hairpin sequences. 3. RESULTS AND DISCUSSIONS 3.1. One-class SVM performance We experimentally evaluated our method by using the three-fold cross-validation procedure as described in Sec- tion 2.1. In order to avoid miRNA hairpins in the same group (defined in [44]) being divided into different folds, we placed all similar miRNA hairpins in the same fold. Three criteria of precision, recall, and F1-measure were used to evaluate the results. We carried out experiments with three types of kernels (linear, polynomial, and radial basic function (RBF)). For each cross-validation run, we used default parameters , δ ,dand various values of pa- rameter v in the range of [0.07, 0.11]. The prediction results are shown in Table 2. It can be seen that one-class SVMs worked well with v= 0.10 and RBF kernels ( γ = 0.0001); the highest F1-measure =95.27%, precision = 94.63%, and recall = 95.92%. The work in this paper is an extension of our conference paper [37]. Basically, there is one improvement here: we tried to check the con- tribution of each kind of features to the prediction results. Table 2. The prediction results of one-class SVMs on the testing dataset. Pre., Rec., and F1. are precision recall and F1-measure, re- spectively. Linear kernel Polynomial kernel RBF kernel v Pre. Rec. F1. Pre. Rec. F1. Pre. Rec. F1. 0.07 88.02 98.66 93.04 88.02 98.66 93.04 91.20 97.32 94.16 0.08 89.09 98.66 93.63 89.09 98.66 93.63 94.67 95.30 94.98 0.09 91.03 95.30 93.11 91.03 95.30 93.11 95.27 94.63 94.95 0.10 93.75 90.60 92.15 93.71 89.93 91.78 95.92 94.63 95.27 0.11 94.37 89.93 92.10 93.71 89.93 91.78 95.24 93.96 94.59 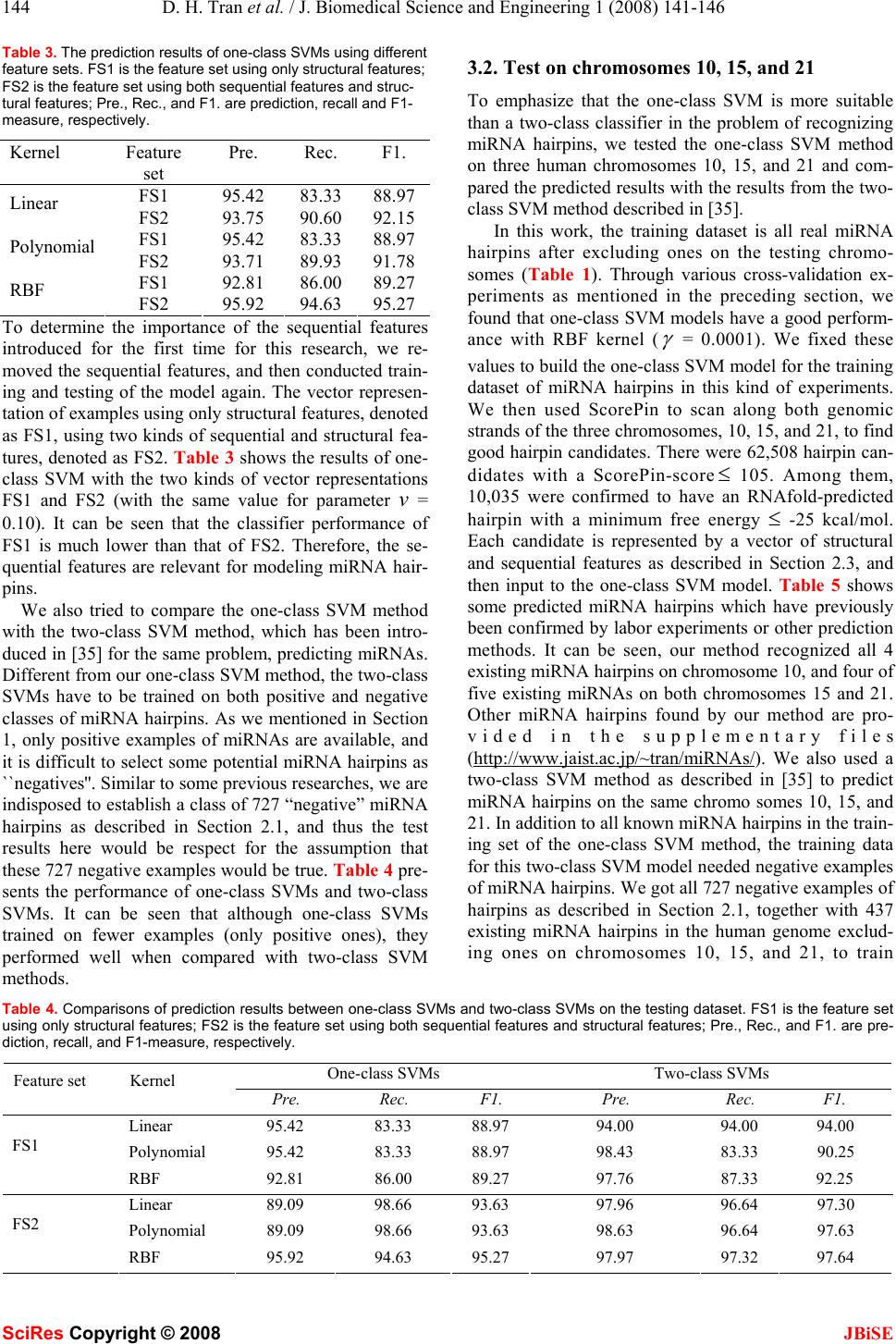 144 D. H. Tran et al. / J. Biomedical Science and Engineering 1 (2008) 141-146 SciRes Copyright © 2008 JBiSE Table 3. The prediction results of one-class SVMs using different feature sets. FS1 is the feature set using only structural features; FS2 is the feature set using both sequential features and struc- tural features; Pre., Rec., and F1. are prediction, recall and F1- measure, respectively. Kernel Feature set Pre. Rec. F1. FS1 95.42 83.33 88.97 Linear FS2 93.75 90.60 92.15 FS1 95.42 83.33 88.97 Polynomial FS2 93.71 89.93 91.78 FS1 92.81 86.00 89.27 RBF FS2 95.92 94.63 95.27 To determine the importance of the sequential features introduced for the first time for this research, we re- moved the sequential features, and then conducted train- ing and testing of the model again. The vector represen- tation of examples using only structural features, denoted as FS1, using two kinds of sequential and structural fea- tures, denoted as FS2. Table 3 shows the results of one- class SVM with the two kinds of vector representations FS1 and FS2 (with the same value for parameter v = 0.10). It can be seen that the classifier performance of FS1 is much lower than that of FS2. Therefore, the se- quential features are relevant for modeling miRNA hair- pins. We also tried to compare the one-class SVM method with the two-class SVM method, which has been intro- duced in [35] for the same problem, predicting miRNAs. Different from our one-class SVM method, the two-class SVMs have to be trained on both positive and negative classes of miRNA hairpins. As we mentioned in Section 1, only positive examples of miRNAs are available, and it is difficult to select some potential miRNA hairpins as ``negatives''. Similar to some previous researches, we are indisposed to establish a class of 727 “negative” miRNA hairpins as described in Section 2.1, and thus the test results here would be respect for the assumption that these 727 negative examples would be true. Table 4 pre- sents the performance of one-class SVMs and two-class SVMs. It can be seen that although one-class SVMs trained on fewer examples (only positive ones), they performed well when compared with two-class SVM methods. 3.2. Test on chromosomes 10, 15, and 21 To emphasize that the one-class SVM is more suitable than a two-class classifier in the problem of recognizing miRNA hairpins, we tested the one-class SVM method on three human chromosomes 10, 15, and 21 and com- pared the predicted results with the results from the two- class SVM method described in [35]. In this work, the training dataset is all real miRNA hairpins after excluding ones on the testing chromo- somes (Table 1). Through various cross-validation ex- periments as mentioned in the preceding section, we found that one-class SVM models have a good perform- ance with RBF kernel ( γ = 0.0001). We fixed these values to build th e one-class SVM model for the training dataset of miRNA hairpins in this kind of experiments. We then used ScorePin to scan along both genomic strands of the three chromosomes, 10, 15, and 21, to find good hairpin candidates. There were 62,508 hairpin can- didates with a ScorePin-score ≤ 105. Among them, 10,035 were confirmed to have an RNAfold-predicted hairpin with a minimum free energy ≤ -25 kcal/mol. Each candidate is represented by a vector of structural and sequential features as described in Section 2.3, and then input to the one-class SVM model. Table 5 shows some predicted miRNA hairpins which have previously been confirmed by labor experiments or other prediction methods. It can be seen, our method recognized all 4 existing miRNA hairpins on chromosome 10, and four of five existing miRNAs on both chromosomes 15 and 21. Other miRNA hairpins found by our method are pro- vided in the supplementary files (http://www.jaist.ac.jp/~tran/miRNAs/). We also used a two-class SVM method as described in [35] to predict miRNA hairpins on the same chromo somes 10, 15, and 21. In addition to all known miRNA hairpins in the train- ing set of the one-class SVM method, the training data for this two-class SVM model needed negative examples of miRNA hairpins. We got all 727 negative examples of hairpins as described in Section 2.1, together with 437 existing miRNA hairpins in the human genome exclud- ing ones on chromosomes 10, 15, and 21, to train Table 4. Comparisons of prediction results between one-class SVMs and two-class SVMs on the testing dataset. FS1 is the feature set using only structural features; FS2 is the feature set using both sequential features and structural features; Pre., Rec., and F1. are pre- diction, recall, and F1-measure, respectively. One-class SVMs Two-class SVMs Feature set Kernel Pre. Rec. F1. Pre. Rec. F1. Linear 95.42 83.33 88.97 94.00 94.00 94.00 Polynomial 95.42 83.33 88.97 98.43 83.33 90.25 FS1 RBF 92.81 86.00 89.27 97.76 87.33 92.25 Linear 89.09 98.66 93.63 97.96 96.64 97.30 Polynomial 89.09 98.66 93.63 98.63 96.64 97.63 FS2 RBF 95.92 94.63 95.27 97.97 97.32 97.64  D. H. Tran et al. / J. Biomedical Science and Engineering 1 (2008) 141-146 145 SciRes Copyright © 2008 JBiSE Table 5. The known miRNA hairpins predicted by one-class SVMs on chromosomes 10, 15, and 21. Location consists of the start point and end point of the miRNA hairpin on the chromo- some. MFE is a minimum free energy of the miRNA hairpin struc- ture. Chr # Location miRNA_ID MFE 17927110:17927200 hsa-mir-511-1 -34.6 17927110:17927200 hsa-mir-511-2 -34.6 52729335:52729425 hsa-mir-605 -54.8 10 104186251:104186341 hsa-mir-146b -41.2 60903206:60903296 hsa-mir-190 -32.5 86956075:86956165 hsa-mir-7-2 -43.1 77289181:77289271 hsa-mir-184 -37.9 15 87712251:87712341 hsa-mir-9-3 -41.1 16833274:16833364 hsa-mir-99a -47.0 25868151:25868241 hsa-mir-155 -39.5 36014883:36014973 hsa-mir-802 -35.0 21 16834016:16834106 hsa-let-7c -43.2 Table 6. The known miRNA hairpins predicted by two-class SVMs on chromosomes 10, 15, and 21. Location consists of the start point and end point of the miRNA hairpin on the chromosome. MFE is a minimum value of the miRNA hairpin structure. Ch # Location miRNA_ID MFE 17927110:17927200 hsa-mir-511-1 -34.6 17927110:17927200 hsa-mir-511-2 -34.6 52729335:52729425 hsa-mir-605 -54.8 10 104186251:104186341 hsa-mir-146b -41.2 60903206:60903296 hsa-mir-190 -32.5 15 86956075:86956165 hsa-mir-7-2 -43.1 16833274:16833364 hsa-mir-99a -47.0 25868151:25868241 hsa-mir-155 -39.5 21 36014883:36014973 hsa-mir-802 -35.0 the discriminative two-class SVM model. Table 6 shows some miRNA hairpins predicted by the two-class SVM model. Among them, all four miRNA hairpins on chro- mosome 10 were identified as same as using the one-class SVM. Consistent with the results reported in [35], the two-class SVM also recognized three of five existing miRNA hairpins on chromosome 21, and two of four on chromosome 15. Especially, while one-class SVM recog- nized correctly an additional miRNA hairpin on chromo- some 21, the two-class SVM predicted them as negatives. The reasons why two-class SVM method incorrectly rec- ognized some known miRNA hairpins might be that the two-class SVM training is based on some negative exam- ples of miRNA hairpins, which might not be true due to the way to select "negative" ones. 4. CONCLUSIONS We have introduced a one-class learning method to pre- dict pre-miRNAs in the human genome. Our one-class support vector machine method has an advantage over other two-class discriminative models: it uses only avail- able positive examples of miRNA hairpins for building the model, while all existing methods for the same prob- lem must use additional negative ones, which are not available, since it is hard to find true negatives for the training of a two-class classifier. Our method showed good performance, and we have illustrated the case of testing on chromosomes 10, 15 and 21, in which our method gave the prediction results more precise than those from an existing two-class support vector machine method. ACKNOWLEDGMENTS The research described in this paper was partially supported by the Insti- tute for Bioinformatics Research and Development of the Japan Science and Technology Agency, and by COE project JCP KS1 of the Japan Advanced Institute of Science and Technology. The first author has been supported by Japanese government scholarship (Monbukagakusho) to study in Japan. The authors also would like to thank Prof. Ivo Hofacker from University of Vienna for providing the ViennaRNA package and Dr. Chih-Jen Lin from National Taiwan University for providing the LIBSVM tool. REFERENCES [1] V. Ambros. (2004) The functions of animal microRNAs. Nature, 431, 350–355. [2] D. P. Bartel. (2004) MicroRNAs: genomics, biogenesis, mechanism, and function. Cell, 116, 281–297. [3] I. Bentwich. (2005) Prediction and validation of microRNAs and their targets. FEBS Lett, 579, 5904–5910. [4] E. Berezikov, E. Cuppen, and R. H. Plas terk. (2006) Approaches to microRNA discovery. Nat. Genet., 38, S2-S7. [5] C. J. Burges. (1998) A tutorial on support vector machines for pat- tern recognition. J. Data Mining and Knowledge Discovery, 2, 121-167. [6] M. T. Bohnsack, K. Czaplinski and D. Grlich. (2004) Exportin 5 is a RanGTP-dependent dsRNA-binding protein that mediates nuclear export of pre-miRNAs. RNA, 10, 185–191. [7] J. Brown, P. Sanseau. (2005) A computational view of microRNAs and their targets. Drug discovery today: biosilico, 10(8), 595–601. [8] C. -C. Chang, and C. -J. Lin. (2001) LIBSVM: a library for support vector machines. [9] Y. Chen, X. Zhou, and T. S. Huang. (2001) One-class SVM for learning in image retrieval. Proc. IEEE Int’l Conf. on Image Processin g, Thessaloniki, Greece. [10] M. A. Denli, B. J. Tops, H. A. Plasterk, R. F. Ketting, and G. J.Hannon. (2004) Processing of primary microRNAs by the Microproc- essor complex. Nature, 432, 231–235. [11] Y. Grad, J. Aach, G. D. Hayes, B. J. Reinhart, G. M. Church, G. Ruvkun, and J. Kim. (2003) Computational and experimental identifica- tion of C. elegans microRNAs. Mol Cell, 11,1253–1263. [12] R. I. Gregory, K. P. Yan, G. Amuthan, T. Chendrimada, B. Dorato- taj, N. Cooch, and R. Shiekhattar. (2004) The microprocessor complex mediates the genesis of microRNAs. Nature, 423, 235–240. [13] S. Griffiths-Jones, R. J. Grocock, S. Dongen, A. Bateman, A. J. Enright. (2006) miRBase: microRNA sequences, targets and gene no- menclature. Nucleic Acids Res., 34, D140–D144. [14] S. Griffiths-Jones. (2004) The microRNA Registry, Nucleic Acids Res., 32, D109–D111. [15] S. A. Helvik, O. S. Jr, and P. Strom. (2007) Reliable prediction of Drosha processing sites improves microRNA gene prediction. Bioinfor-  146 D. H. Tran et al. / J. Biomedical Science and Engineering 1 (2008) 141-146 SciRes Copyright © 2008 JBiSE matics, 23(2), 142-149. [16] I. L. Hofacker, S. Fontana, W. Stadler, S. Bonhoeffer, M. Tacker, and P. Schuster. (1994) Fast folding and comparison of RNA secondary structures, Monatshefte f. Chem ie, 125, 167-188. [17] I. L. Hofacker. (2003) Vienna RNA secondary structure server. Nucleic Acids Res, 31, 3429–3431. [18] M. Kiriakidou, P. T. Nelson, A. Kouranov, P. Fitziev, C. Bouyiou- kos, Z. Mourelatos, and A. Hatzigeorgiou. (2004) A combined computa- tional experimental approach predicts human microRNA targets. Genes Dev, 18, 1165–1178. [19] A. Kowalczyk, and B. Raskutti. (2002) One-class svm for yeast regulation pr ediction. Proc. SI GKDD Explorations Workshop, 99–100. [20] R. Kohavi. (1995) A study of cross-validation and bootstrap for accuracy estimation and model selection. Proc. 14th IJCAI, San Fran- cisco, CA, Morgan Kaufmann Pubshers, 113 7–1143,. [21] Y. Kong, J.-H. Han. (2005) MicroRNA: Biological and computa- tional perspective. Geno. Prot. Bioinfo., 3(2), 62–72. [22] J. Krol, K. Sobczak, U. Wilcztnska, M. Drath, A. Jasinska, D. Kaczynska, and W. J. Krzyzosiak. (2004) Structural features of mi- croRNA (miRNA) precursors and their relevance to miRNA biogenesis and small interfering RNA/short hairpin RNA design. J Biol Chem, 279, 42230–42239. [23] M. Lagos-Quintana, R. Rauhut, W. L endeckel, and T. Tuschl. (2001) Identification of novel gene coding for small expressed RNAs. Science, 294, 853–858. [24] E. C. Lai, P. Tomancak, R. W. Williams, and G. M. Rubin. (2003) Computational identification of Drosophila microRNA genes. Genome Biol, 4, R42. [25] Y. Lee, C. Ahn, J. Han, H. Choi, J. Yim, P. Provost, O. Radmark, S. Kim, and V. N. Kim. (2003) The nuclear RNase III Drosha initiates microRNA processing. Nature, 424, 415–419. [26] Y. Lee, M. Kim, J. Han, K. Yeom, S. H. Lee, S. H. Baek, and V. N. Kim. (2004) MicroRNA genes are transcribed by RNA polymerase II. EmboJ, 23,4051–4060. [27] L. P. Lim, M. E. Glasner, S. Yekta, C. B. Burge, and D. P. Bartel. (2003) Vertebrate microRNA genes. Science, 299, 1540. [28] L. P. Lim, N. C. Lau, E. G. Weinstein, A. Abdelhakim, S. Yekta, M. W. Rhoades, C. B. Burge, and D. P. Bartel. (2003) The microRNAs of Caenorhabditis elegans. Genes Dev, 17, 991-1008. [29] E. Lund, S. Guttinger, A. Calado, J. E. Dahlberg, and U. Kutay. (2004) Nuclear export of microRNA precursors. Science, 303, 95–98. [30] L. M. Manevitz, and M. Yousef. (2001) One-class SVMs for docu- ment classification. Journal of Machine Learning, 2, 139–154. [31] J. W. Nam, K. R. Shin, Y. V. Lee, N. Kim, and B. T. Zhang. (2005) Human microRNA prediction through a probabilistic co-learning model of sequence and structure. Nucleic Acids Res., 33, 3570–3581. [32] U. Ohler, S. Yekta, L. P. Lim, D. P. Bartel, and C. B. Burge. (2004) Patterns of flanking sequence conservation and a characteristic upstream motif for microRNA gene identification. RNA, 10, 1309–1322. [33] B. Scholkopf, J. C. Platt, J. Shawe-Taylor, A. J. Smola, and R. C. Williamson. (2001) Estimating the support of a high-dimensional distri- bution. Neural Comput, 13, 1443–1471. [34] P. Strom, O. S. Jr, M. Nedland, T. B. Grnfeld, Y. Lin, M. B. Bass, J. Canon. (2006) Conserved microRNA characteristics in mam- mals. Oligonucleotides, 16, 115–144. [35] K. Szafranski, M. Megraw, M. Reczko, G. H. Hatzigeorgiou. (2006) Support vector machine for predicting microRNA hairpins. Proc. The 2006 International Conference on Bioinformatics and Computational Biology, 270–276. [36] A. Tsirigos, and I. Rigoutsos. (2005) A sensitive, support-vector- machine method for the detection of horizontal gene transfers in viral, archaeal and bacterial genomes. Nucleic Acids Research, 33(12):3699– 3707. [37] D. H. Tran, T. H. Pham, K. Satou, and T. B. Ho. (2008) Prediction of microRNA hairpins using one-class support vector machine. Proc. The 2nd international conference on bioinformatics and biomedical engineering (iCBBE), Sanghai, China, May 16-18. [38] V. Vapnik. Statistical learning theory, Wiley, Chichester, United Kingdom, 19 98. [39] X. Xie, J. Lu, E. J. Kulbokas, T. R. Golub, V. Mootha, K. Lindblad- Toh, E. S. Lander, and M. Kellis. (2005) Systematic discovery of regula- tory motifs in human promoters and 3’ UTRs by comparison of several mammals. Nature, 434, 338–346. [40] C. Xue, F. Li, T. He, G. P. Liu, Y. Li, and X. Zhang. (2005) Classi- fication of real and pseudo microRNA precursors using local structure- sequence features and support vector machine. BMC Bioinformatics, 6, 310. [41] L. H. Yang, W. Hsu, M. L. Lee, and L. Wong. (2006) Identification of microRNA precursors via SVM, Proc. The 4th Asia- PacificBioinformatics Conference, 267–276. [42] Y. Zeng, R. Yi, and B. R. Cullen. (2005) Recognition and cleavage of primary microRNA precursors by the nuclear processing enzyme Drosha. Embo J, 24,138–148. [43] http://www.csie.ntu.edu.tw/ cjlin/libsvm/ [44] http://microrna.sanger.ac.uk/sequences/index.shtml [45] http://www.tbi.univie.ac.at/RNA/ |