 America n Journal of Analy tic al Chemistry, 2011, 2, 352-362 doi:10.4236/ajac.2011. 23043 Published Online July 2011 (http://www.scirp.org/journal/ajac) Copyright © 2011 SciRes. AJAC A Validated Liquid Chromatography-Mass Spectromet ry Method for the Detection and Quantification of Oxidative Metabolites of 2,2',4, 4'-Tetrabromodiphenyl Ether in Rat Hepatic Micro som es Sarah Catherine Moffatt1, Patrick R obert Edwards2, András Szeitz1, Stelvio Mario Bandiera1* 1Faculty of Pharmaceutical Sciences, The University of British Colum bia, Vancouver, Canada 2Faculty of Pharmacy, University of Toronto, Toronto, Canada E-mail: sarah.moffatt@gmail.com, bandiera@interchange.ubc.ca Received January 5, 2011; revised March 4, 2011; accepted March 15, 2011 Abstract In the present study, we developed and validated an analytical method using ultra performance liquid chro- matography-mass spectrometry (UPLC/MS) for the quantitative determination of 2,2',4,4'-tetrabromodiphenyl ether (BDE-47) metabolism by rat hepatic microsomes. BDE-47 is a brominated flame retardant that was widely used in a variety of consumer products and has subsequently been identified as a ubiquitous environmental contaminant. Hydroxy-bromodiphenyl ethers (OH-BDEs) were isolated from rat hepatic microsomes by liquid-liquid extraction. Chromatographic separation was achieved by UPLC on a C18 column with gradient elution using a mobile phase consisting of methanol and water, each containing 0.1% formic acid, at a flow rate of 0.2 mL/min. Detection and quantification were performed using a mass spectrometer in single ion recording mode with negative electrospray ionization. The UPLC/MS method was validated for linearity, limit of quantification (LOQ), accuracy, precision and recovery. The weighted cali- bration curves (1/X2) were linear over a concentration range of 5 - 250 nM with LOQ values between 5 nM and 5 0 nM for t he ind ivid ual OH -BDEs. Intra - and inter- day accuracy (%DEV) and precision (%RSD ) val- ues ranged from –11.7% to 9.5% and 5.9% to 16.5%, respectively. Recovery values of 70% to 90% were obtained for all OH-BDEs. The validated method allowed us to successfully analyze metabolite formation following incubation of BDE-47 with hepatic microsomes prepared from phenobarbital-treated rats. Results demonstrate that the UPLC/MS method has sufficient sensitivity and reproducibility to fully characterize t he in vitro metabolism of BDE-47 and possibly other PBDEs. Keywords: BDE-47, Hepatic Metabolism, Polybrominated Diphenyl Ethers, Rat Hepat ic Mic rosom es, Ultra Performance Liquid Chromatography-Mass Spectrometry. 1. Introduction Polybrominated diphenyl ethers (PBDEs) are haloge- nated aromatic hydrocarbons that have been used as ad- ditive flame retardants on a variety of consumer products since 1965 [1]. PBDEs were marketed as commercial mixtures containing a limited number of the 209 possible brominated diphenyl ether (BDE) congeners [2]. The penta-BDE mixture, which was used extensively in North America [3], was composed predominantly of 2,2', 4,4'-tetrabromodiphenyl ether (BDE-47), 2,2',4,4',5-pen- tabromodiphenyl ether (BDE-99), 2,2',4,4',6-pentabromodiphenyl ether (BDE-100), 2,2',4,4',5,5'-hexabromo - diphenyl ether (BDE-153) and 2,2',4,4',5,6'-hexabromo- diphenyl ether (BDE-154) [4]. Penta-BDE was applied to epoxy resins, textiles, paints and flexible polyurethane foam, which was used in up- holstered furniture, mattresses and carpet padding [5,6]. PBDEs are not chemically bound to the polymer com- ponents of the products to which they are applied and can be released into the environment during manufacture [5], use [7] and disposal [8] of these products. This fac- tor, together with the high lipophilicity, chemical stabil- ity and the widespread use of BDE mixtures has resulted  S. C. MOFFATT ET AL. Copyright © 2011 SciRes. AJAC in the ubiquitous distribution of PBDEs in the environ- ment [1]. BDE-47, for example, has been detected in air [9], sediment [10], fish [10,11], marine mammals [12,13] and in human blood [14], adipose tissue [15] and breast milk [16] and is frequently the predominant PBDE con- gener found in biotic samples [1]. Studies with labora- tor y animal s have sho wn tha t de velop mental expo sure to BDE-47 caused alterations in neuromotor activity [17] and e xposur e in utero produced changes to the reproduc- tive system and thyroid gland of female rat pups [18]. Laboratory studies have shown that PBDEs can be metabolized by hepatic cytochrome P450 enzymes to hydroxy-BDEs (OH-BDEs) [19-22]. OH-BDEs are of toxicological interest as OH-BDEs show a greater affin- ity for the thyroid hormone receptor than the natural li- gand or PBDEs themselves [19]. OH-BDEs have been detected in blood and feces of rodents treated with BDE-47 [21] and various OH-BDEs have been found in human plasma [23]. Several studies, including those mentioned above, examined the formation of OH-BDEs; however, the specific enzymes and enzyme kinetics of OH-BDEs formation are poorly understood. Characteri- zation of BDE-47 metabolism in vitro is needed to de- velop a better understanding of the role of metabolism in the bioaccumula tion and toxicity of BDE -47. The most common method for the detection and quan- tification of PBDEs and OH-PBDEs in environmental samples has been gas chromatography-mass spectrome- try (GC/MS) or gas chromatography coupled with elec- tron capture detection (GC/ECD) [19]. In a study that examined the endocrine disrupting activity of BDEs fol- lowing hepatic biotransformation, Hamers et al. [19] identified six hydroxylated metabolites of BDE-47 using a GC/MS method. GC-based methods are sensitive, but require derivatization of OH-BDEs, additional sample preparation time, the use of harmful derivatizing agents and possible underestimation of OH -BDE concentrations due to incomplete der ivatization. Liquid chro matograph y coupled with mass spectrometry (LC/MS) provides an alternative analytical technique that does not require de- rivatization. Mas et al. d e monstrated that LC/MS can be used to detect and quantify OH -BDEs in soi l, fish, sludge and particulate matter that was spiked with a mixture of OH-BDEs [24]. However, their LC/MS method was not validated in a biological matrix [24] and its applicability to the detection and quantification of oxidative metabo- lites of BDE-47 generated in vitro or in vivo is unknown. The aim of the present study was to develop and vali- date a UPLC/ MS-based analytical method to detect and quantify OH-BDEs and apply this method to investigate the in vitro biotransformation of BDE-47 by rat hepatic microsomes. 2. Materials and Methods 2.1. Chemicals and Reagents BDE-47 (neat, 99% purity) was obtained from Chiron (Trondheim, Norway). 4'-Hydroxy-2,2',4-tribromodi- phenyl ether (4'-OH-BDE-17), 2'-hydroxy-2,4,4'-tribro- modiphenyl ether (2'-OH-BDE-28), 4-hydroxy-2,2',3,4'- tetrabromodiphenyl ether (4-OH-BDE-42), 3-hydroxy-2, 2',4,4'-tetrabromodiphenyl ether (3-OH-BDE -47), 5-hy- droxy-2,2',4,4'-tetrabromodiphenyl ether (5-OH-BDE- 47), 6-hydroxy-2,2',4,4'-tetrabromodiphenyl ether (6- OH-BDE-47), 4'-hydroxy-2,2',4,5'-tetrabromodiphenyl ether (4'-OH-BDE-49) (10 µg/mL or 50 µg/mL in aceto- nitrile, purity of at least 98%) and 4'-hydroxy-2,2',4,6'- tetrachlorobiphenyl (4'-OH-CB-50, neat, 99% purity) were purchased from AccuStandard (New Haven, Con- necticut, USA). Magnesium chloride, sucrose and nico- tinamide ade nine dinucleotide p hosphate (NADPH) were purchased from Sigma-Aldrich (Oakville, Ontario, Can- ada). Methanol, methyl-tert-butyl ether, hexane, isopro- panol, sodium hydroxide, hydrochloric acid and mono- and di-basic potassium phosphate were purchased from Fisher Scientific (Ottawa, Ontario, Canada). Hydroch- loric acid and all organic solvents were HPLC- grade or higher. Ultra pure water was obtained using a Milli-Q Synthesis syst em (Millipore, Billerica, MA, USA). 2.2. Rat Hepati c Micro so mes Adult male Long Evans rats (body weight between 160- 190 g) were purchased from Charles River Laboratories (Montreal, PQ, Canada). Rats were cared for in accor- dance with the principles and guidelines outlined by the Canadian Council of Animal Care. Rats ( ) were treated with sodium phenobarbital (PB, 80 mg/kg/day) for 3 days, as previously described by Edwards et al. [25]. Pooled hepatic microsomes were prepared by dif- ferential centrifugation as previously described [26]. He- patic microsomes were aliquoted and stored at –80˚C until use. Protein concentration was determined by the method of Lowry, et al. with bovine serum albumin as the standard [27]. 2.3. Standard Solutions A stock solution of BDE-47 at a concentration of 2.5 mM was prepared in methanol. A stock solution of OH-BDE standards containing 4'-OH-BDE-17, 2'-OH-BDE-28, 4-OH-BDE-42, 3-OH-BDE-47, 5-OH-BDE-47, 6- OH-BDE-47 and 4'-OH-BDE-49 (at 1.25 µM each) was prepared in methanol. A second 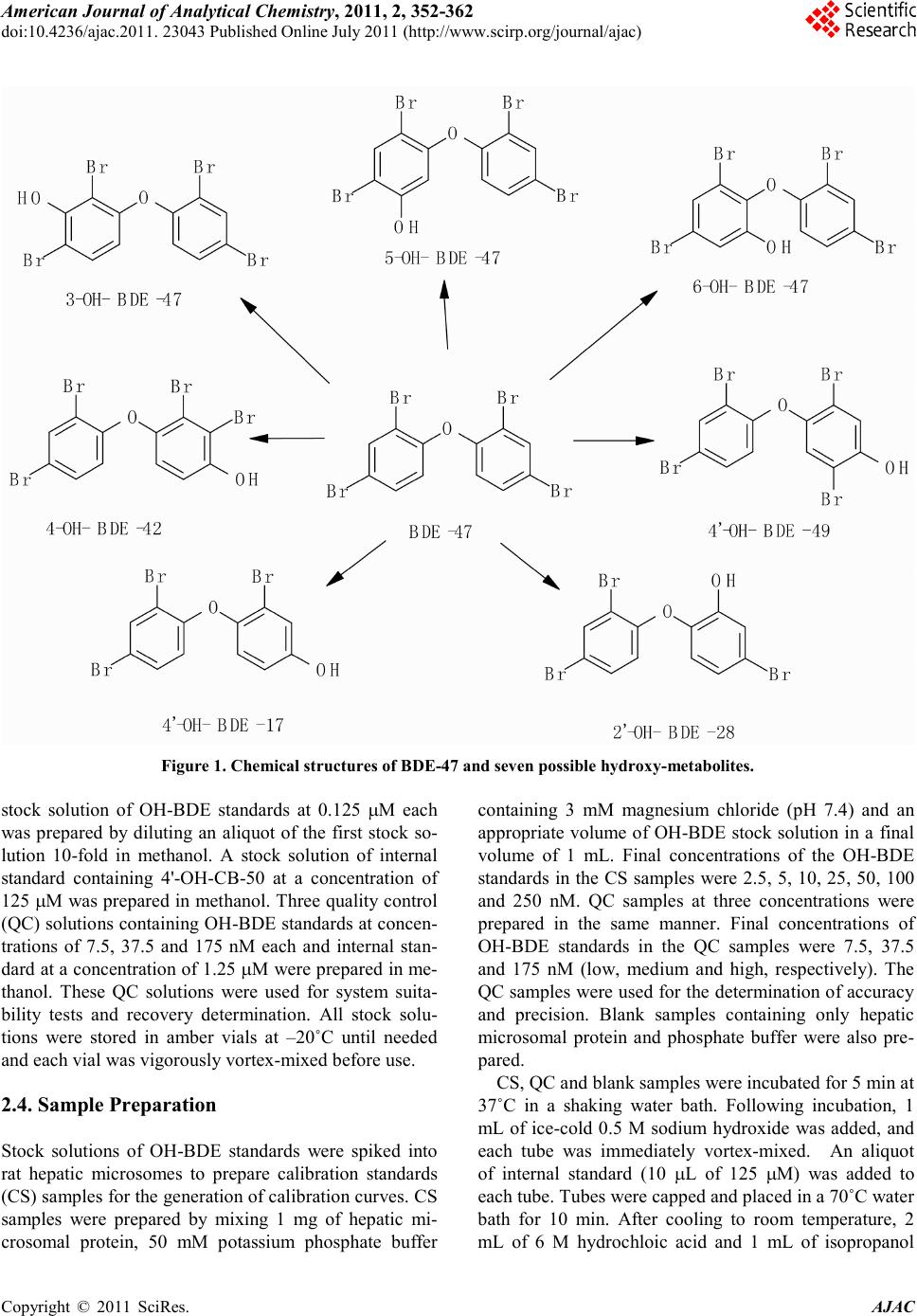 America n Journal of Analy tic al Chemistry, 2011, 2, 352-362 doi:10.4236/ajac.2011. 23043 Published Online July 2011 (http://www.scirp.org/journal/ajac) Copyright © 2011 SciRes. AJAC Figure 1 . Chemical stru ct ures of BDE-47 a nd seven possibl e hydroxy-metabolites. stock solution of OH-BDE standards at 0.125 µM each was prepared by diluting an aliquot of the first stock so- lution 10-fold in methanol. A stock solution of internal standard containing 4'-OH-CB-50 at a concentration of 125 µM wa s pr epared in methanol. Three quality control (QC) solutio ns co ntaini ng OH -BDE standards at concen- trations of 7.5, 37.5 and 175 nM each and internal stan- dard at a concentration of 1.25 µM were prepared in me- thanol. These QC solutions were used for system suita- bility tests and recovery determination. All stock solu- tions were stored in amber vials at –20˚C until needed and eac h via l was vi go ro u sl y vor te x -mixed before use. 2.4. Sample Preparation Stock solutions of OH-BDE standards were spiked into rat hepatic microsomes to prepare calibration standards (CS) samples for the generation of calibration curves. CS samples were prepared by mixing 1 mg of hepatic mi- crosomal protein, 50 mM potassium phosphate buffer containing 3 mM magnesium chloride (pH 7.4) and an appropriate volume of OH-BDE stock solution in a fina l volume of 1 mL. Final concentrations of the OH-BDE standards in the CS samples were 2.5, 5, 10, 25, 50, 100 and 250 nM. QC samples at three concentrations were prepared in the same manner. Final concentrations of OH-BDE standards in the QC samples were 7.5, 37.5 and 175 nM (low, medium and high, respectively). The QC samples were used for the deter mination of accuracy and precision. Blank samples containing only hepatic microsomal protein and phosphate buffer were also pre- pared. CS, QC and blank samples were incubated for 5 min at 37˚C in a shaking water bath. Following incubation, 1 mL of ice-cold 0.5 M sodium hydroxide was added, and each tube was immediately vortex-mixed. An aliquot of internal standard (10 µL of 125 µM) was added to each tube. Tubes were capped and placed in a 70˚C water bath for 10 min. After cooling to room temperature, 2 mL of 6 M hydrochloic acid and 1 mL of isopropanol 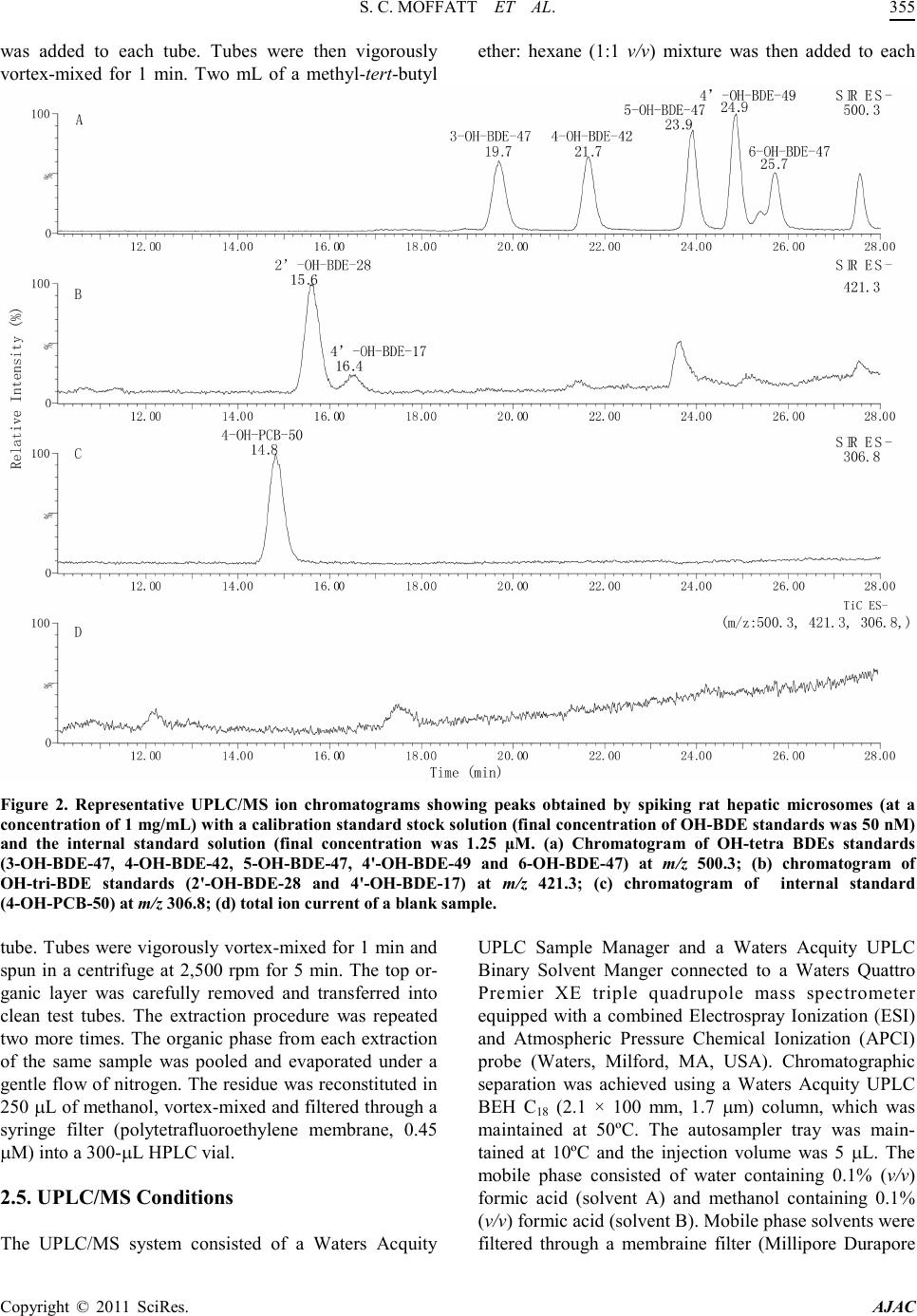 S. C. MOFFATT ET AL. Copyright © 2011 SciRes. AJAC was added to each tube. Tubes were then vigorously vortex-mixed for 1 min. Two mL of a methyl-tert-butyl ether: hexane (1:1 v/v) mixture was then added to each Figure 2. Representative UPLC/MS ion chromatograms showing peaks obtained by spiking rat hepatic microsomes (at a conce ntr ati on of 1 mg/ mL) w ith a cali brat ion sta ndar d st oc k sol utio n (f inal co nce ntr atio n of OH-B DE standards w as 50 nM) and the internal standard solution (final concentration was 1.25 µM. (a) Chromatogram of OH-tetra BDEs standards (3-OH-BDE-47, 4-OH-BDE-42, 5-OH-BDE-47, 4'-OH-BDE-49 and 6-OH-B DE-47) at m/z 500.3; (b) chromatogram of OH-tri-BDE standards (2'-OH-BDE-28 and 4'-OH-BDE-17) at m/z 421.3; (c) chromatogram of internal standard (4-OH-PCB-50) at m/z 306.8; (d) total ion current of a blank sample. tube. Tubes were vi gorousl y vor tex-mixed for 1 min and spun in a centrifuge at 2,500 rpm for 5 min. The top or- ganic layer was carefully removed and transferred into clean test tubes. The extraction procedure was repeated two more times. The organic phase from each extraction of the same sample was pooled and evaporated under a gentl e flo w of nit rogen. The res idue wa s reconsti tuted in 250 µL of met hano l, vort ex-mi xed and filtere d thro ugh a syringe filter (polytetrafluoroethylene membrane, 0.45 µM) into a 300-µL HPLC v ial. 2.5. UPLC/MS Conditions The UPLC/MS system consisted of a Waters Acquity UPLC Sample Manager and a Waters Acquity UPLC Binary Solvent Manger connected to a Waters Quattro Premier XE triple quadrupole mass spectrometer equipped with a combined Electrospray Ionization (ESI) and Atmospheric Pressure Chemical Ionization (APCI) probe (Waters, Milford, MA, USA). Chromatographic separation was achieved using a Waters Acquity UPLC BEH C18 (2.1 × 100 mm, 1.7 µm) column, which was maintained at 50ºC. The autosampler tray was main- tained at 10ºC and the injection volume was 5 µL. The mobile phase consisted of water containing 0.1% (v/v) formic acid (solvent A) and methanol containing 0.1% (v/v) formic acid (solvent B). Mob ile phase solvents were filtered through a membraine filter (Millipore Durapore 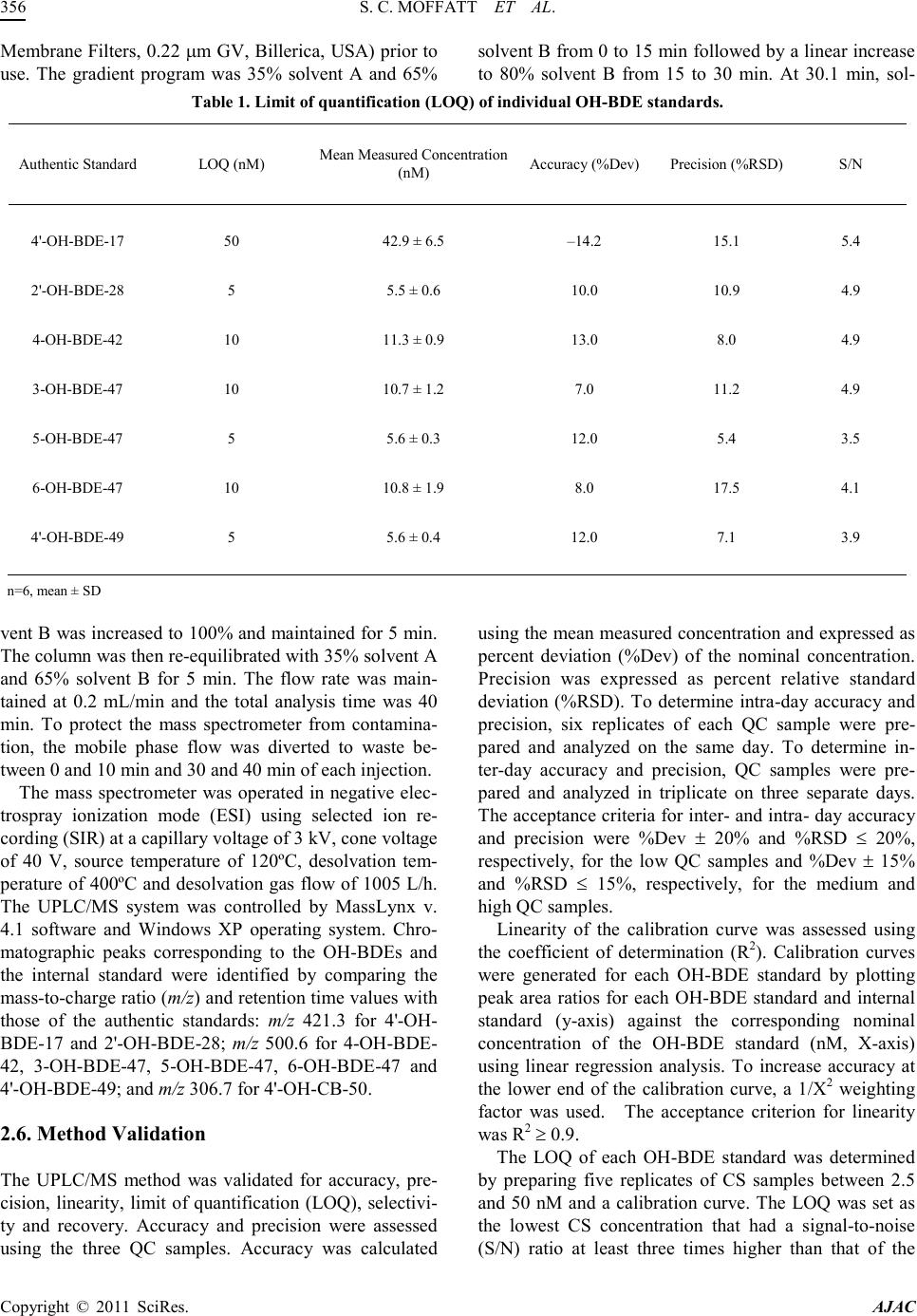 S. C. MOFFATT ET AL. Copyright © 2011 SciRes. AJAC Membrane Filters, 0.22 µm GV, B illerica, USA) prior to use. The gradient program was 35% solvent A and 65% solvent B from 0 to 15 min followed by a linear increase to 80% solvent B from 15 to 30 min. At 30.1 min, sol- Table 1. Limit of quantification (LOQ) of individu al OH-BDE standards. Authentic Standard LOQ (nM) Mea n Measured Concentrati on (nM) Accuracy (%Dev) Precisi on (%RSD) S/N 4'-OH-BDE-17 50 42.9 ± 6.5 –14.2 15.1 5 .4 2'-OH-BDE-28 5 5.5 ± 0.6 10.0 10.9 4.9 4-OH-BDE-42 10 11.3 ± 0.9 13.0 8.0 4.9 3-OH-BDE-47 10 10.7 ± 1.2 7.0 11.2 4.9 5-OH-BDE-47 5 5.6 ± 0.3 12.0 5.4 3.5 6-OH-BDE-47 10 10.8 ± 1.9 8.0 17.5 4.1 4'-OH-BDE-49 5 5.6 ± 0.4 12.0 7.1 3.9 n=6, mean ± SD vent B was increased to 100% and maintained for 5 min. The co lu mn wa s t he n r e-equilibrated with 35% solvent A and 65% solvent B for 5 min. The flow rate was main- tained at 0.2 mL/min and the total analysis time was 40 min. To protect the mass spectrometer from contamina- tion, the mobile phase flow was diverted to waste be- tween 0 and 10 min and 30 and 40 min of each injection. The mass spectrometer was operated in negative elec- trospray ionization mode (ESI) using selected ion re- cording (SIR) at a capillary voltage of 3 kV, cone voltage of 40 V, source temperature of 120ºC, desolvation tem- perature of 400ºC and desolvation gas flow of 1005 L/h. The UPLC/MS system was controlled by MassLynx v. 4.1 software and Windows XP operating system. Chro- matographic peaks corresponding to the OH-BDEs and the internal standard were identified by comparing the mass -to -c harge ratio (m/z) and re tentio n ti me va lue s wi th those of the authentic standards: m/z 421.3 for 4'-OH- BDE-17 and 2'-OH-BDE-28; m/z 500.6 for 4-OH-BDE- 42, 3-OH-BDE-47, 5-OH-BDE-47, 6-OH-BDE-47 and 4'-OH-BDE-49; and m/z 306.7 for 4'-OH-CB-50. 2.6. Method Validation The UPLC/MS method was validated for accuracy, pre- cision, linearity, limit of quantification (LOQ), selectivi- ty and recovery. Accuracy and precision were assessed using the three QC samples. Accuracy was calculated using the mean measured concentration and expressed as percent deviation (%Dev) of the nominal concentration. Precision was expressed as percent relative standard deviation (%RSD). To determine intra-day accuracy and precision, six replicates of each QC sample were pre- pared and analyzed on the same day. To determine in- ter-day accuracy and precision, QC samples were pre- pared and analyzed in triplicate on three separate days. The acceptance criteria for inter- and i ntra - day accurac y and precision were %Dev ± 20% and %RSD ≤ 20%, respectively, for the low QC samples and %Dev ± 15% and %RSD ≤ 15%, respectively, for the medium and high Q C sample s . Linearity of the calibration curve was assessed using the coefficient of determination (R2). Calibration curves were generated for each OH-BDE standard by plotting peak area ratios for each OH-BDE standard and internal standard (y-axis) against the corresponding nominal concentration of the OH-BDE standard (nM, X-axis) using linear regression analysis. To increase accuracy at the lower end of the calibration curve, a 1/X2 weighting factor was used. The acceptance criterion for linearity was R2 ≥ 0.9. The LOQ of each OH-BDE standard was determined by preparing five replicates of CS samples between 2.5 and 50 nM and a calibration curve. The LOQ was set as the lowest CS concentration that had a signal-to-noise (S/N) ratio at least three times higher than that of the  S. C. MOFFATT ET AL. Copyright © 2011 SciRes. AJAC blank sample with a ± 20% Dev and ≤ 20% RSD. The S/N ratio was determined with MassLynx using the peak-to-peak method. Selectivity was assessed by visually comparing the Table 2. I ntra-day accurac y (%Dev) and precis ion (%RS D). Metabolite Nominal Concentration (nM) Mean Measured Concentratio n (nM) Accuracy (%Dev) Precision (%RSD) 4'-OH-BDE-17 QC-Low (7.5) < LOQ n. d. n. d. QC-Med (37.5) < LO Q n. d. n. d. QC-High (175) 167. 0 ± 1 7 .7 –4.6 10.6 2'-OH-BDE-28 QC-Low (7.5) n. d. n. d. n. d. QC-Med (37.5) 36.1 ± 3.3 –3.7 9 .1 QC-High (175) 162. 3 ± 1 5 .7 –7.3 9 .7 4-OH-BDE-42 QC-Low (7.5) < LOQ n. d. n. d. QC-Med (37.5) 36.7 ± 2.2 –2.1 5 .9 QC-High (175) 153. 9 ± 1 5 .4 –12.1 10.0 3-OH-BDE-47 QC-Low (7.5) < LOQ n. d. n. d. QC-Med (37.5) 36.8 ± 2.8 –2.1 7 .6 QC-High (175) 156. 2 ± 1 4 .9 –10.7 9.5 5-OH-BDE-47 QC-Low (7.5) 8.2 ± 1 .4 9.3 17.0 QC-Med (37.5) 36.6 ± 3.0 –2.4 8.2 QC-High (175) 155. 7 ± 1 5 .3 –11.0 9 .8 6-OH-BDE-47 QC-Low (7.5) < LOQ n. d. n. d. QC-Med (37.5) 38.3 ± 3.0 2.1 7.8 QC-High (175) 178. 2 ± 1 9 .1 1.8 10.7 4'-OH-BDE-49 QC-Low (7.5) 7.8 ± 0 .5 4.0 6.4 QC-Med (37.5) 35.8 ± 3.2 –4.5 8 .9 QC-High (175) 157. 3 ± 1 3 .9 –10.1 8.8 < LOQ = Below limit of quantificati on; n .d., not determined because metabolite concent ration was below the LOQ; n = 6, mean ± SD. chromatograms obtained from blank samples with chro- mato grams fro m spiked CS samples at the LOQ value of each OH-BDE standard. Chromatograms were examined for the presence of interfering peaks with retention time s that overlapped those of OH-BDE standard s or the inter- nal standard. Recovery rates were determined using QC solutions and QC samples. Recovery was calculated by comparing the peak area of each OH-BDE standard in the QC sam- ple with that in the QC solution at the same concentra- tion. 2.7. In Vitro Biotransformat ion of BDE-47 Following validation, t he UPLC/MS method was applied to investigate the in vitro biotransformation of BDE-47. For mation of hydroxy BDE-47 metabolites by rat hepati c microsomes was determined using a reaction mixture containing 50 mM phosphate buffer, 0 - 2 mg/mL of rat hepatic miroso mal prote in and 50 µM BDE-47 (20 µL of 2.5 mM i n met ha no l) i n a fi nal vo lu me o f 0 .9 9 mL. A fter a 5-mi n pr e-incubation at room temperature, the reaction was initiated by addition of 10 µL of 100 mM NADPH  S. C. MOFFATT ET AL. Copyright © 2011 SciRes. AJAC (final concentration 1 mM) and allowed to proceed at 37˚C in a shaking water bath for 0 to 30 min. The reac- tion was terminated by the addition of 1 mL of ice-cold 0.5 M sodium hydroxide. Extraction and quantification of the OH -BDE metabolites was performed as described above. Samples were prepared in duplicate for each as- Table 3. I nter-day accur acy (%Dev) and precisio n (%RSD). Metabolite Nominal Concentration (nM) Mean Measured Concentration (nM) Accuracy (%Dev) Precision (%RSD) 4'-OH-BDE-17 QC-Low (7.5) < LOQ n. d. n. d. QC-Med (37.5) < LOQ n. d. n. d. QC-High (175) 164. 3 ± 2 2 .3 –6.1 13.6 2'-OH-BDE-28 QC-Low (7.5) n. d. n. d. n. d. QC-Med (37.5) 37.3 ± 4.8 –0.4 12.7 QC-High (175) 158. 4 ± 1 2 .0 –9.5 7 .6 4-OH-BDE-42 QC-Low (7.5) < LOQ n. d. n. d. QC-Med (37.5) 37.8 ± 4.0 0.8 10.6 QC-High (175) 155. 4 ± 1 1 .5 –11 .2 7.4 3-OH-BDE-47 QC-Low (7.5) < LOQ n. d. n. d. QC-Med (37.5) 37.6 ±3 .0 0.3 10.1 QC-High (175) 155. 2 ± 1 1 .5 –11 .3 7.4 5-OH-BDE-47 QC-Low (7.5) 6.9 ±2.5 –8.0 8.0 QC-Med (37.5) 37.4 ±4 .2 –0.3 11.3 QC-High (175) 156. 3 ± 1 3 .0 –10.7 8.3 6-OH-BDE-47 QC-Low (7.5) < LOQ n. d. n. d. QC-Med (37.5) 39.5 ± 4.3 5.3 10.9 QC-High (175) 182. 8 ± 1 4 .8 4.4 8.1 4'-OH-BDE-49 QC-Low (7.5) 7.7 ± 0 .5 2.7 6.5 QC-Med (37.5) 35.7 ± 4.8 –4.8 13.4 QC-High (175) 154. 6 ± 1 1 .0 –11 .7 7.1 < LOQ = Below limit of quantifi cation; n. d., no t deter mined becau se m e t a bo lite concentr ation wa s be l o w the L O Q ; n = 12 (Day 1 n = 6, Day 2 n = 3, Day 3 n. = 3) me an ± SD. say and experiments were performed 3 times on separate days. Blank samples contained only rat hepatic micro- somes and buffer. Negative control samples did not con- tain BDE-47, NADPH, or rat hepatic microsomes. QC samples were included in each experiment to assess me- thod performance. 3. Results and Discussion 3.1. Optimization of UPLC/MS Parameters APCI and ESI in negative and positive modes were compared for the detection of OH-BDE standards by mass spectrometry using SIR. OH-BDE standards were not detected in positive ion mode. ESI operated in nega- tive electrospray ion mode produced larger peak area counts than APCI. BDE-47 was not detected with APCI 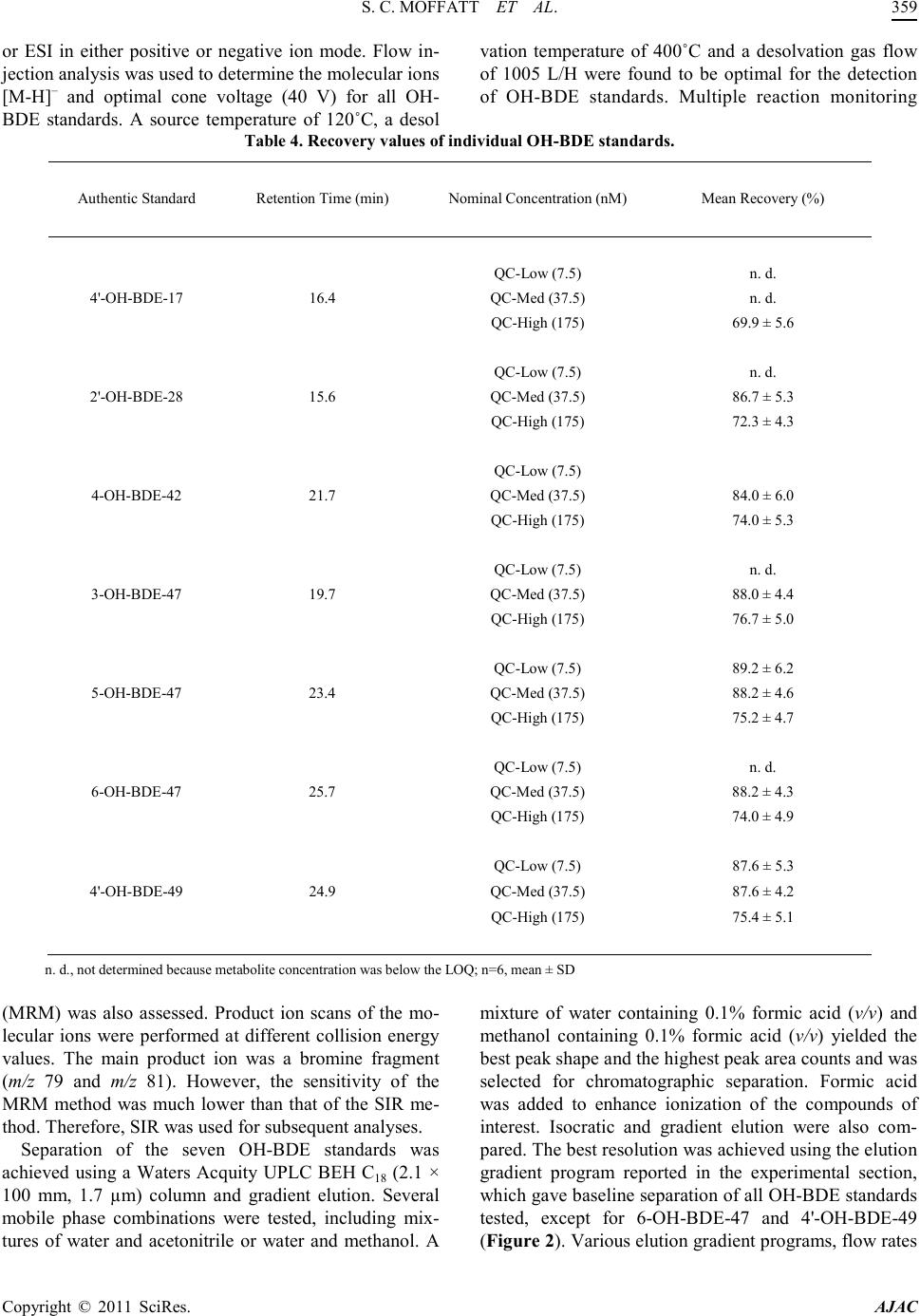 S. C. MOFFATT ET AL. Copyright © 2011 SciRes. AJAC or ESI in either positive or negative ion mode. Flow in- jection analysis was used to determine the molecular ions [M-H]– and optimal cone voltage (40 V) for all OH- BDE standards. A source temperature of 120˚C, a desol vation temperature of 400˚C and a desolvation gas flow of 1005 L/H were found to be optimal for the detection of OH-BDE standards. Multiple reaction monitoring Table 4. Recovery value s of individual OH -BDE stan da rds. Authentic Standard Retention Time (min) Nominal Concentration (nM) Mea n Recovery (%) 4'-OH-BDE-17 QC-Low (7.5) n. d. 16.4 QC-Med (37.5) n. d. QC-High (175) 69.9 ± 5.6 2'-OH-BDE-28 QC-Low (7.5) n. d. 15.6 QC-Med (37.5) 86.7 ± 5.3 QC-High (175) 72.3 ± 4.3 4-OH-BDE-42 QC-Low (7.5) 21.7 QC-Med (37.5) 84.0 ± 6.0 QC-High (175) 74.0 ± 5.3 3-OH-BDE-47 QC-Low (7.5) n. d. 19.7 QC-Med (37.5) 88.0 ± 4.4 QC-High (175) 76.7 ± 5.0 5-OH-BDE-47 QC-Low (7.5) 89.2 ± 6.2 23.4 QC-Med (37.5) 88.2 ± 4.6 QC-High (175) 75.2 ± 4.7 6-OH-BDE-47 QC-Low (7.5) n. d. 25.7 QC-Med (37.5) 88.2 ± 4.3 QC-High (175) 74.0 ± 4.9 4'-OH-BDE-49 QC-Low (7.5) 87.6 ± 5.3 24.9 QC-Med (37.5) 87.6 ± 4.2 QC-High (175) 75.4 ± 5.1 n. d., not determined becaus e metabolite concentration was below th e LOQ; n=6, mean ± SD (MRM) was also assessed. Product ion scans of the mo- lecular ions were performed at different collision energy values. The main product ion was a bromine fragment (m/z 79 and m/z 81). However, the sensitivity of the MRM method was much lower than that of the SIR me- thod. There fore, SIR was used for subse que nt analyses. Separation of the seven OH-BDE standards was achieved using a Waters Acquity UPLC BEH C18 (2.1 × 100 mm, 1.7 µm) column and gradient elution. Several mobile phase combinations were tested, including mix- tures of water and acetonitrile or water and methanol. A mixture of water containing 0.1% formic acid (v/v) and methanol containing 0.1% formic acid (v/v) yielded the best peak shape and the highest peak area counts and was selected for chromatographic separation. Formic acid was added to enhance ionization of the compounds of interest. Isocratic and gradient elution were also com- pared. The best resolution was achieved using the elution gradient program reported in the experimental section, which gave baseline separation of all OH-BDE standards tested, except for 6-OH-BDE-47 and 4'-OH-BDE-49 (Figure 2). Various elution gra dient pr ogra ms, flow rates 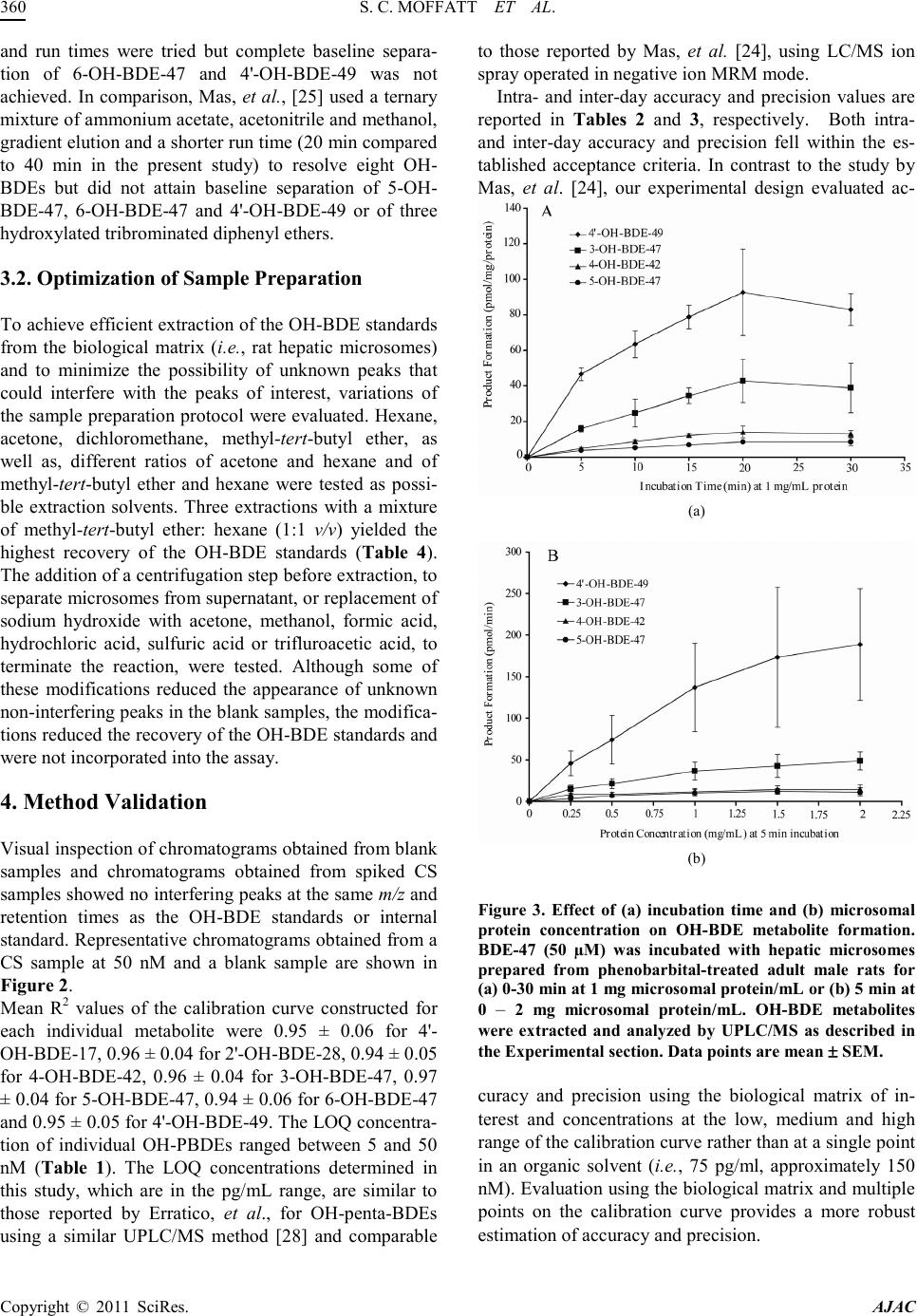 S. C. MOFFATT ET AL. Copyright © 2011 SciRes. AJAC and run times were tried but complete baseline separa- tion of 6-OH-BDE -47 and 4'-OH-BDE-49 was not achieved. In comparison, Mas, et al., [25] used a ternary mixtur e of a m monium aceta te , aceto nitrile a nd methano l, gradient elution and a shorter run time (20 min compared to 40 min in the present study) to resolve eight OH- BDEs but did not attain baseline separation of 5-OH- BDE-47, 6-OH-BDE-47 and 4'-OH-BDE-49 or of three hydroxylated tribrominated diphenyl ethers. 3.2. Optimization of Sample Preparation To achieve efficient extraction of the OH-BDE standards from the biological matrix (i.e., rat hepatic microsomes) and to minimize the possibility of unknown peaks that could interfere with the peaks of interest, variations of the sample preparation protocol were evaluated. Hexane, acetone, dichloromethane, me th yl-tert-butyl ether, as well as, different ratios of acetone and hexane and of me th yl-tert-butyl ether and hexane were tested as possi- ble extraction solvents. Three extractions with a mixture of methyl-tert -butyl ether: hexane (1:1 v/v) yielded the highest recovery of the OH-BDE standards (Table 4). The add ition of a ce ntrif ugati on step b efore e xtractio n, to separate microsomes from supernatant, or replacement of sodium hydroxide with acetone, methanol, formic acid, hydrochloric acid, sulfuric acid or trifluroacetic acid, to terminate the reaction, were tested. Although some of these modifications reduced the appearance of unknown non-interfe ri ng pea ks in t he b lank sa mpl es, t he modi fica- tions reduced the recovery of the OH-BDE standards and were not incorporated into the assay. 4. Method Validation Visual inspection of chromatograms obtained from blank samples and chromatograms obtained from spiked CS samples s ho wed no interfering peaks at the same m/z and retention times as the OH-BDE standards or internal standard. Representative chromatograms obtained from a CS sample at 50 nM and a blank sample are shown in Figure 2. Mean R2 values of the calibration curve constructed for each individual metabolite were 0.95 ± 0.06 for 4'- OH-BDE-17, 0.96 ± 0.04 for 2'-OH-BDE-28, 0.94 ± 0.05 for 4-OH-BDE-42, 0.96 ± 0.04 for 3-OH-BDE-47, 0.97 ± 0.04 for 5-OH-BDE-47, 0.94 ± 0.06 for 6-OH-BDE-47 and 0.95 ± 0.05 for 4'-OH-BDE-49. The LOQ concentra- tion of individual OH-PBDEs ranged between 5 and 50 nM (Table 1). The LOQ concentrations determined in this study, which are in the pg/mL range, are similar to those reported by Erratico, et al., for OH-penta-BDEs using a similar UPLC/MS method [28] and comparable to those reported by Mas, et al. [24], using LC/MS ion spray operated in negative ion MRM mode. Intra- and inter-day accuracy and precision values are reported in Tables 2 and 3, respectively. Both intra- and inter-day accuracy and precision fell within the es- tablished acceptance criteria. In contrast to the study by Mas, et al. [24], our experimental design evaluated ac- (a) (b) Figure 3. Effect of (a) incubation time and (b) microsomal protein concentration on OH-BDE metabolite formation. BDE-47 (50 µM) was incubated with hepatic microsomes prepared from phenobarbital-treated adult male rats for (a) 0-30 min at 1 mg microsomal protei n/ mL or (b) 5 min a t 0 – 2 mg microsomal protein/mL. OH-BDE metabolites were extracted and analyzed by UPLC/MS as described in the Exper imental section. Data point s are mea n ± SEM. curacy and precision using the biological matrix of in- terest and concentrations at the low, medium and high range of the calibration curve rather than at a single point in an organic solvent (i.e., 75 pg/ml, approximately 150 nM). Evaluati on usi ng t he biological matri x and multiple points on the calibration curve provides a more robust estimation of accuracy and precision.  S. C. MOFFATT ET AL. Copyright © 2011 SciRes. AJAC Recovery values for the low and medium QC sample ranged between 84% and 89% for all OH-BDEs. Recov- ery values for the high QC sample were slightly lower, ranging between 70% and 77% (Table 4). The recovery values are higher than those reported in a previous s tud y that used GC-based methods [19], but lower than those reported by Erratico, et a l. [28] using a similar method to identify the oxida tive metabolites of BDE -99. Increasing the extraction volume or the number of extractions, did not increase recovery rates of the OH-PBDE standard s. The method validation studies confirm that OH-BDEs are amenable to direct analysis by UPLC/MS in negat ive electrospray ion mode without the need for derivatization or extensive fragme ntation and that the UPLC/MS-based analytical method that we developed can be used for the separation, detection a nd quantification of OH-BDEs in a complex biological matrix such as rat liver microsomes. 4.1. Biotr an s for mation of BDE-47 by Rat Liver Microsom es The validated UPLC/MS method was applied to analyze the oxidative metabolism of BDE-47 in vitro. Incubation of BDE-47 with rat hepatic microsomes prepared from PB-treated adult male rats produced five OH-tetra-BDE metabolites (4-OH-BDE-42, 3-OH-BDE-47, 5-OH-BDE- 47, 6-OH-BDE-47 and 4'-OH-BDE-49) which were de- tected and identified by their retention times and values. The major metabolite was 4'-OH-BDE-49. There was no evidence for the fo rmation of 4'-OH-BDE-17 or 2'- OH-BDE-28 by hepatic microsomes prepared from PB- treated rats. No other metabolite or unidentified peaks were observed. The effect of varying incubation time (0 - 30 min) and protein concentration (0 - 2 mg/mL) on formation of the five hydroxylated metabolites was investigated using a substrate (BDE-47) concentration of 50 µM. Formation of 4 -OH-BDE-42, 3-OH-BDE-47, 5-OH-BDE-47 and 4'- OH-BDE-49 was linear for the first 5 min of incubation at a microsomal protein concentration of 1 mg/mL (Fig- ure 3a) and approximately linear up to a microsomal protein concentration of 1 mg/mL at an incubation time of 5 min (Figure 3b). Under the e xperi mental conditio ns used, formation of 6-OH-BDE-47 was detected but could not be quantified because the concentration of this me- tabolite remained below the LOQ value (10 nM). 5. Conclusions A UP LC/M S-based analytical method for the separation, detection and quantification of seven possible oxidative metabolites of BDE-47 in rat liver microsomes was de- veloped and validated. The method was applied to ana- lyze the oxidative metabolism of BDE-47 in vitro and allowed us to d etect formation of five monohydroxylated metabolites o f BDE-47 and quantify formation of four of the metabolites. Our method represents an improvement of previously published LC/MS methods because valida- tion was performed using the biological matrix of inter- est and at the low, medium and high ends of the calibra- tion curve providing a more complete assessment of the accuracy, precision, recovery and LOQ of the analytical method. Quantification of hydroxylated metabolites of BDE-47 generated by hepatic microsomes following a 5 min incubation at 1 mg microsomal protein/mL demon- strates the sensitivity and feasibility of the method for further in vitro metabolic stud ies, includ ing reaction phe - notyping, analyzing enzyme-catalyzed reaction kinetics and enzyme inhibition. The selectivity and reproducibil- ity of the UPLC/MS method combined with the in vitro metabolism assay will be useful for measuring metabol- ism of BDE-47 in human liver samples and in liver preparations from additional species. 6. References [1] P. Vonderheide, K. E. Mueller, J. Meij a and G. L. Welsh, “Polybrominated Diphenyl Ethers: Causes for Concern and Knowledge Gaps Regarding Environmental Distribu- tion, Fate and Toxicity,” Science of the Total Environ- ment, Vol. 400, No. 1-3, 2008, pp. 425-436. doi:10.1016/j.scitotenv.2008.05.003 [2] M. Alaee, P. Arias, A. Sjödin and Å. Bergman, “An Overview of Commercially Used Brominated Flame Re- tardants, Their Applications, Their Use Patterns in Dif- ferent Countries/Regions and Possible Modes of Re- lease,” Environment International, Vol. 29, No. 6, 2003, pp. 683-689. doi:10.1016/S0160-4120(03)00121-1 [3] R. C. Hale, M. Alaee, J. B. Manchester-Neesvig, H. M. Stapleton and M. G. Ikonomou, “Polybrominated Di- phenyl Ether Flame Retardants in the North American Environment,” Environment International, Vol. 29, No. 6, 2003, pp. 771-779. doi:10.1016/S0160-4120(03)00113-2 [4] M. J. La Guardia, R. C. Hale and E. Harvey, “Detailed Polybrominated Diphenyl Ether (PBDE) Congener Composition of the Widely Used Penta-, Octa-, and De- ca-PBDE Technical Fame-Retardant Mixtures,” Envi- ronmental Science and Technology, Vol. 40, No. 20, 2006 , p p. 6247-6254. doi:10.1021/es060630m [5] R. C. Hale, M. J. La Guardia, E. Harvey and T. M. Mai- nor, “Potential Role of Fire Retardant-Treated Polyure- Thane Foam as a Source of Brominated Diphenyl Ethers to the US Environment,” Chemosp here, Vol. 46, No. 5, 2002 , p p. 729-735. doi:10.1016/S0045-6535(01)00237-5 [6] F. Rahman, K. H. Langford, M. D. Scrimshaw and J. N. Lester, “Polybrominated Diphenyl Ether (PBDE) Flame Retardants,” Science of the Total Environment, Vol. 275, No. 1-3, 2001, pp. 1-17.  S. C. MOFFATT ET AL. Copyright © 2011 SciRes. AJAC doi:10.1016/S0048-9697(01)00852-X [7] H. M. Stapleton, A. Sjödin, R. S. Jones, S. Niehüser, Y. Zhang and D. G. Patterson, “Serum Levels of Polybro- Minated Diphenyl Ethers (PBDEs) in Foam Recyclers and Carpet Installers Working in the United States,” En- vironmental Science and Technology, Vol. 42, No. 9, 2008 , p p. 3453-3458. doi:10.1021/es7028813 [8] D. O. Odusanya, J. O. Okonkwo, and B. Botha, “Poly- Brominated Diphenyl Ethers (P BDEs) in Leachates From Selected Landfill Sites in South Africa,” Waste Manage- ment, Vol. 29, No. 1, 2009, pp. 96-102. [9] M. Noël, N. Dangerfield, R. A. S. Hourston, W. Belzer, P. Shaw, M. B. Yunker and P. S. Ross. “Do trans-Pacific Air Masses Deliver PBDEs to Coastal British Columbia, Canada?” Environmental Pollution, Vol. 157, No. 12, 2009 , p p. 3404-3412. doi:10.1016/j.envpol.2009.06.025 [10] K. Law, T. Halldorson, R. Danell, G. Stern, S. Gewurtz, M. Alaee, C. Marvin, M. Whittle, and G. Tomy, “Bioac- Cumulation and Trophic Transfer of Some Brominated Flame Retardants in a Lake Winnipeg (Canada) Food Web,” Environmental Toxicology and Chemistry, Vol. 25, No. 8, 2006, pp. 2177-2186. doi:10.1897/05-500R.1 [11] N. G. Dodder, B. Strandberg and R. A. Hites, “Concen- trations and Spatial Variations of Polybrominated Di- phenyl Eth ers and Several Organochlorine Compounds in Fishes From the Northeastern United States,” Envi ron- mental Science and Technology, Vol. 36, No. 2, 2002, pp. 146-151. doi:10.1021/es010947g [12] M. A. McKinney, I. Stirling, N. J. Lunn, E. Peacock and R. J. Letcher, “The Role of Diet on Long-Term Concen- tration and Pattern Trends of Brominated and Chlorinated Contaminants in Western Hudson Bay Polar Bears, 1991-2007,” The Science of the Total Environment, Vol. 408, No. 24, 2010, pp. 6210-6222. doi:10.1016/j.scitotenv.2010.08.033 [13] C. Kelly, M. G. Ikonomou, J. D. Blair and F.A.P.C. Go- bas, “Hydroxylated and Methoxylated Polybrominated Diphenyl Ethers in a Canadian Arctic Marine Food Web,” Environmental Science and Technology, Vol. 42, No. 19, 200 8, pp. 7069-7077. doi:10.1021/es801275d [14] A. Mazdai, N. G. Dodder, M. P. Abernathy, R. A. Hites and R. M. Bigsby, “Polybrominated Diphenyl Ethers in Maternal and Fetal Blood Sa mp l es,” Environmental Health Perspectives, Vol. 111, No. 9, 2003, pp. 1249- 1252. doi:10.1289/ehp.6146 [15] B. John son -Rest repo , K. Kann an, D.P. Rap apor t and B.D. Rodan, “Polybrominated Diphenyl Ethers and PolyChlo- rinated Biphenyls in Human Adipose Tissue from New York,” Environmental Science and Technology, Vol. 39, No. 14, 2005, pp. 5177-5182. doi:10.1021/es050399x [16] A. Schecter, M. Pavuk, O. Päpke, J. J. Ryan, L. Birnbaum and R. Rosen, “Polybrominated Diphenyl Ethers (PBDEs) in U.S. Mothers' Milk,” Environmental Health Perspectives, Vol. 111, No. 14, 2003, pp. 1723-1729. doi:10.1289/ehp.6466 [17] J. R. Gee and V . C. Moser, “Acut e Postnatal Exposu re to Brominated Diphenylether 47 Delays Neuromotor Onto- geny and Alters Motor Activity in Mice,” Neurotoxicol- ogy and Treatment, Vol. 30, No. 2, 2008, pp. 79-87. [18] C. E. Talsness, S. N. Kuriyama, A. Sterner-Kock, P. Schnitker, S. W. Grande, M. Shakibaei, A. Andrade, K. Grote and I. Chahoud, “In Utero and Lactational Expo- sures to Low Doses of Polybrominated Diphenyl Ether- 47 Alter the Reproductive System and Thyroid Gland of Female Rat Offspring,” Environmental Health Perspec- tives, Vol. 116, No. 3, 2008, pp. 308-314. doi:10.1289/ehp.10536 [19] T. Hamers, J. H. Kamstra, E. Sonneveld, A. J. Murk, T. J. Visser, M. J. M. Van Velzen, A. Brouwer and Å. Berg- man, “Biotransformation of Brominated Flame Retar- dants into Potentially Endocrine-Disrupting Metabolites, with Special Attention to 2,2',4,4'-Tetrabromodiphenyl ether (BDE-47),” Molecular Nutrition & Food Research, Vol. 52, No. 2, 2008 , pp. 284-298. doi:10.1289/ehp.10536 [20] J. M. Sanders, L.-J. Chen, E. H. Lebetkin and L. T. Bur- ka, “Metabolism and Disposition of 2,2',4,4'-Tetrabromodi- phenyl Ether Following Ad mi n i s- tration of Single or Multiple Doses to Rats and Mice,” Xenobiotica, Vol. 36, No. 1, 2006, pp. 103-117. doi:10.1080/00498250500485107 [21] G. Marsh, M. Athanasiadou, I. Athanassiadis and A. Sandholm, “Identification of Hydroxylated Metabolites in 2,2',4,4'-Tetrabromodiphenyl Ether Exposed Rats ,” Che- mosphere, Vol. 63, No. 4 , 2006, pp. 690-697. doi:10.1016/j.chemosphere.2005.07.072 [22] S. W. Cheng, K. Randal and, A. T. Kotchevar. “In Vitro Metabolism Studies of Polybrominated Diphenyl Ethers using Rat and Human Liver Microsomes,” American Journal of Biochemistry and Biotechnology, Vol. 4, No. 3, 2008, pp . 295-303. doi:10.3844/ajbbsp.2008.295.303 [23] M. Athanasiadou, S. N. Cuadra, G. Marsh, Å. Bergman and K. Jakobsson, “Polybrominated Diphenyl Ethers (PBDEs) and Bioaccumulative Hydroxylated PBDE Me- tabolites in Young Humans from Managua, Nicaragua,” Environmental Health Perspectives, Vol. 116, No. 3, 2008, pp. 400-408. doi:10.1289/ehp.10713 [24] S. Mas, O. Jauregui, F. Rubio, A. de Juan, R. Tauler and S. Lacorte, “Comprehensive Liquid Chromatogra- phy-Ionspray Tandem Mass Spectrometry Method for the Identification and Quantification of Eight Hydroxylated Brominated Diphenyl Eth ers in Environmental Matrices,” Journal of Mass Spectro metry, Vol. 42, No. 7, 2007, pp. 890-900. doi:10.1002/jms.1224 [25] P. R. Edwards, E. G. Hrycay and S.M. Bandiera, “DifFe- rential Inhibition of Hepatic Microsomal AlkoxyreSoru- fin O-Dealkylation Activities by Tetrachlorobiphenyls, ” Chemico-Biological Interactions, Vol. 169, No. 1 2007, pp. 42-52. doi:10.1016/j.cbi.2007.05.004 [26] T. Omura and R. Sato, “The Carbon Monoxide-Binding Pigment of Liver Microsomes I: Evidence for its Hemo- protein Nature,” The Journal of Biological Chemistry, Vol. 239, No. 7, 1964, pp. 2370-2378. [27] O. H. Lowry, N. J. Rosebrough, A. L. Farr and R. J. Randall, “Protein Measurement with the Folin Phenol Reagent,” The Journal of Biological Chemistry, Vol. 193, 1951 , p p. 265-275.  S. C. MOFFATT ET AL. Copyright © 2011 SciRes. AJAC [28] A. Errat ico, A. Szei tz and S. M. Band iera. “Vali dation of a Novel in Vitro Assay Using Ultra Performance Liquid Chromatography-Mass Spectrometry (UPLC/MS) to Detect and Quantify Hydroxylated Metabolites of BDE-99 in Rat Liver Microsomes,” Journal of chroma- tography B, Analytical technologies in the biomedical and life sciences, Vol. 878, No. 19, 2010, pp. 1562-1568. doi:10.1016/j.jchromb.2010.04.014
|