 Vol.3, No.7, 580-593 (2011) Natural Science http://dx.doi.org/10.4236/ns.2011.37081 Copyright © 2011 SciRes. OPEN ACCESS The common physical origin of the glass transition, macromolecular entanglement and turbulence Jia-Lin Wu College of Material Science and Engineering, Donghua University, Shanghai, China; *Corresponding Author: jlwu@dhu.edu.cn Received 27 March 2011; revised 27 April 2011; accepted 3 May 2011. ABSTRACT The interface excitation (IE) on intermolecular interface is a common concept connecting the glass transition (GT), macromolecular entan- glement (ME), and turbulence. IE has an addi- tional repulsion energy and extra vacancy vol- ume that result from the two neighboring mo- lecules with antiparallel delocalization all in, e.g., the z-axial ground state of single-molecule in- stantaneous polarized dipole at GT. IEs only occur in the 8 orders of 2D IE loop-flows on lo- cal x-y projection plane. Theoretical proof of the 3.4 power law of ME viscosity reveals that 1) the delocalization mode of GT and solid-liquid tran- sition is solitary wave; wave-particle duality of solitary wave is ascribed to the equal probabili- ties between appearing and disappearing of IE loop-flow in inverse cascade and cascade mode; 2) macromolecular chain-length in ME motion corresponds to Reynolds number in hydrody- namics; both the ME motion and the turbulent flow obey the same scale law. IE is not the ex- citation of dipole energy level at GT. However, when IEs are associated with the energy levels of instantaneous polarized dipole, we predict that the coherent structure formed by multilevel 8 orders of 2D IE loop-flows is the physical ori- gin of turbulence based on the universal ran- dom delocalization transition theory. Keywords: Glass Transition; Mosaic Stricture; Reptation; Coherent Structure; Random Transition 1. INTRODUCTION Turbulence is well-deservedly often called as “the last great unsolved problem of the classical physics.” Despite enormous advances in sciences and many highly impor- tant new scientific domains emerged in the past century, the modern status of the turbulence theory is quite ex- ceptional and differs from that of all other new sciences [1]. On the other hand, Nobel laureate Philip Anderson wrote, “The deepest and most interesting unsolved pro- blem in solid state is probably the nature of glass and glass transition (GT)” [2]. In physical theory, many complicated phenomena originate from the global prop- erties (Berry’s Phase) of simple quantum systems [3] and the emergent properties of the many-times repeated ap- plication of quite simple physical laws [4]. It is discov- ered in this paper that the physical origin of turbulence is the same as that of GT and macromolecular entangle- ment (ME) when solving GT and ME in polymer phys- ics. The three famous problems all attribute to the in- corporation of many physical ingredients, containing the maximal Berry’s phase of interface excitation (IE) loop-flows and the slowest also the maximum Brownian regression-order potential in random systems. The ap- pearance of the 8th order IE loop-flow plays the domi- nant role in inducing molecule-cluster localization - de- localization transition and flow speedup along one-di- mensional direction in a reference 3D domain. The minimum energy mode of the structure (confor- mational) rearrangements in supercooled liquid could be deduced from the motion mode of ME in melt liquid state. This means that we can directly use the GT theory (also proof-test the GT theory) to prove the 3.4 power law of viscosity in ME [5,6], which is a well - known experiment law found more than 60 years ago and yet failing to have theoretical proof ever since. This power law is theoretically proved in this paper. The real sur- prise comes when we deduce the scale law of ME mo- tion from the power law and find that the macromolecu- lar chain-length corresponds to the Reynolds number in hydrodynamics; both the ME motion and the turbulent flow obey the same scale law. These findings indicate that both ME and turbulence relate to the same theory and the solid-to-liquid GT is the paradigm from disorder to more disorder transition in inverse cascade – cascade mode. Very recently, Aleiner et al. [7] show theoretically that the one-dimensional gas of short-range interacting ato- mic bosons in the presence of disorder can undergo a  J.-L. Wu / Natural Science 3 (2011) 580-593 Copyright © 2011 SciRes. OPEN ACCESS 581 finite-temperature phase transition between two distinct states: fluid and insulator, and the Anderson localization - delocalization transition can be identified in the dy- namics of expansion of disordered bosonic clouds re- leased from the superimposed trapping potential. This study may also provide an existent evidence for univer- sal random delocalization transitions presented in this paper, if we regard the energy of finite-temperature as the Brownian directional regression vibration energy along one-dimensional direction in a 3D local space and the transition between two distinct states as the inverse cascade—cascade mode of localization—delocalization transition in random systems. This paper is organized as follows. Section 2 reca- pitulates the mosaic structure theory of GT, including the fixed point of self-similar two-body interaction, frustra- tion-percolation transition; and further proves the inverse cascade – cascade mode is the fundamental mode of GT via a validity check of GT theory for the well-known abnormal exponential function in glass state. In Section 3, we unveil the structure of ME by means of the theo- retical proof for the 3.4 power law of viscosity; and show the theoretical contrast between random delocali- zation transition and phase transition. In Section 4, we present the theoretical predictions for the coherent struc- ture of turbulence based on the universal geometric frus- tration-percolation transition theory. 2. MOSAIC STRUCTURE 2.1. Inverse Cascade–Cascade Mode One of the GT theories is the inverse cascade–cascade mode along local one direction (e.g. notated in local z-axial, or on x-y projection plane) in a reference (thaw, excited) domain. The definition of excited domain in GT, similar to that in a ferromagnetic material, is a contigu- ous region in which the direction of thawing mole- cule-cluster spontaneous delocalization (Section 3.3) is uniform and different from that in neighboring regions. This idea comes from the insight for cooperative orien- tation activation energy, ΔEco, on melt high-speed spin- ning-line [8]. When the work of the stress on over 5000 M/min spinning-line reaches ΔEco, the structure of the yarn is stable and reaches full orientation, called as FOY (Full Orientation Yarn) in current polyester fiber industry. This phenomenon is called stress-induced GT. The rate of change of the stress-induced liquid-to-solid GT is 107 times of that in general quencher from melt state to frozen glass state. A logical explanation for melt high-speed spinning is that the macromolecules can complete liquid-to-solid GT with full orientation within the millisecond of time in z-space. Their motion mode, within the entire range from melt transition (MT) tem- perature Tm to GT temperature Tg, allows the direction of inverse cascade – cascade in every excited domain is in arbitrary in melt state and all apt to z-axial on melt spin- ning-line; and finally the mode is frozen in glasses as the soft matrix. At solid-to-liquid GT, increasing tempera- ture only increases the number of IE loop-flows per unit time to augment the number of thawing domains to ac- celerate GT. According to the fundamental view and central assumption of de Gennes’ soft matrix on mosaic structures in glasses [9], an intrinsic 8 orders of transient 2D mosaic geometric structure theory of GT has been proposed [10], Figure 1. The structure is formed by the 8 orders of 2D IE loop- flows from small to large in inverse cascade and rear- rangement structure in cascade. The number of IEs on the 8th order loop is identified by means of the geomet- ric frustration, i.e., the appearance of the 8th order di- rectly corresponds to that of boson peak and geometric frustration in order to satisfy the claims for the theory of GT [9-12]. The number of IE of the 8th order loop equals to that of the 7th order, and the directions of the two loops are opposite. Thus the 8 orders of 2D IE loop- flows with one external degree of freedom (DoF) may be also regarded as a domain-scale interacting classical boson formed by 200 z-component chain-particles (mo- lecules) [10] in a z-axial thaw domain. Furthermore, this GT theory shows explicitly that the geometric frustration also directly corresponds to the geometric frustration- -percolation transition of the 8th order loop-flow to balance the potential and the kinetic [10]. The centric 8th order (V8) loop one by one connects with its neighboring 4 7th order (V7) loops in Figure 1. When the 4 V7 loops, one by one, all evolve to their V8 loops (each V8 loop has one external DoF); the central V8 loop obtains 5 inner DoF and cascades to vanish (that is also the microscopic melting mode with renewed cluster energy, kTm° (v8)). This evolving picture holds true for all IE loops that form the flow-percolation at the GT. In Figure 1, each (small square) chain-particle has a z-axial displacement energy that comes from the Berry’s phase of parallel transports of its 4 IEs surround its z-axis. The 8th order loop induces each of 136 parti- cles to hop in z-axial respectively at different regression times [10]. The central 5 cavity cells, in accordance with de Gennes’ simple picture of “a localized ‘vacancy’ among clusters” [9], are also the mosaic cells denoting the 5 cooperative excited particles [10] delocalized in the correlation of sharing energy Ec (Section 3). This struc- ture can deduce three non-integrable energies: kT2°, kTg°, kTm° and an icosahedral directional ordering at the GT. The 8 orders of IE loop-flows also define the z-axial self-similar 8 orders of transient 2D clusters vi (relaxa- tion time i) and 8 orders of transient 3D hard spheres i 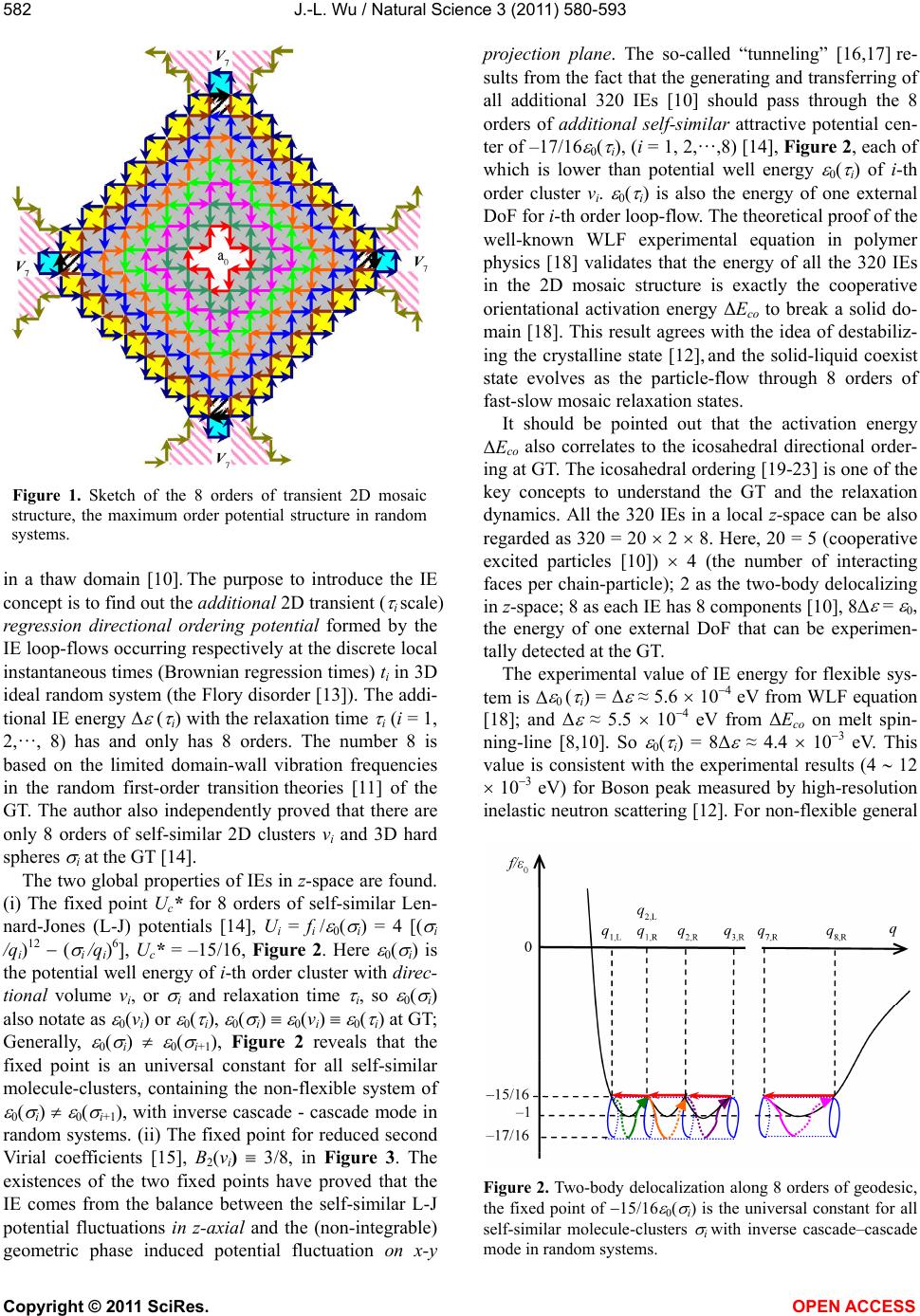 J.-L. Wu / Natural Science 3 (2011) 580-593 Copyright © 2011 SciRes. OPEN ACCESS 582 Figure 1. Sketch of the 8 orders of transient 2D mosaic structure, the maximum order potential structure in random systems. in a thaw domain [10]. The purpose to introduce the IE concept is to find out the additional 2D transient ( i scale) regression directional ordering potential formed by the IE loop-flows occurring respectively at the discrete local instantaneous times (Brownian regression times) ti in 3D ideal random system (the Flory disorder [13]). The addi- tional IE energy Δ ( i) with the relaxation time i (i = 1, 2,…, 8) has and only has 8 orders. The number 8 is based on the limited domain-wall vibration frequencies in the random first-order transition theories [11] of the GT. The author also independently proved that there are only 8 orders of self-similar 2D clusters vi and 3D hard spheres i at the GT [14]. The two global properties of IEs in z-space are found. (i) The fixed point Uc* for 8 orders of self-similar Len- nard-Jones (L-J) potentials [14], Ui = fi / 0( i) = 4 [( i /qi)12 ( i /qi)6], Uc* = –15/16, Figure 2. Here 0( i) is the potential well energy of i-th order cluster with direc- tional volume vi, or i and relaxation time i, so 0( i) also notate as 0(vi) or 0( i), 0( i) 0(vi) 0( i) at GT; Generally, 0( i) 0( i+1), Figure 2 reveals that the fixed point is an universal constant for all self-similar molecule-clusters, containing the non-flexible system of 0( i) 0( i+1), with inverse cascade - cascade mode in random systems. (ii) The fixed point for reduced second Virial coefficients [15], B2(vi) 3/8, in Figure 3. The existences of the two fixed points have proved that the IE comes from the balance between the self-similar L-J potential fluctuations in z-axial and the (non-integrable) geometric phase induced potential fluctuation on x-y projection plane. The so-called “tunneling” [16,17] re- sults from the fact that the generating and transferring of all additional 320 IEs [10] should pass through the 8 orders of additional self-similar attractive potential cen- ter of –17/16 0( i), (i = 1, 2,…,8) [14], Figure 2, each of which is lower than potential well energy 0( i) of i-th order cluster vi. 0( i) is also the energy of one external DoF for i-th order loop-flow. The theoretical proof of the well-known WLF experimental equation in polymer physics [18] validates that the energy of all the 320 IEs in the 2D mosaic structure is exactly the cooperative orientational activation energy ΔEco to break a solid do- main [18]. This result agrees with the idea of destabiliz- ing the crystalline state [12], and the solid-liquid coexist state evolves as the particle-flow through 8 orders of fast-slow mosaic relaxation states. It should be pointed out that the activation energy ΔEco also correlates to the icosahedral directional order- ing at GT. The icosahedral ordering [19-23] is one of the key concepts to understand the GT and the relaxation dynamics. All the 320 IEs in a local z-space can be also regarded as 320 = 20 2 8. Here, 20 = 5 (cooperative excited particles [10]) 4 (the number of interacting faces per chain-particle); 2 as the two-body delocalizing in z-space; 8 as each IE has 8 components [10], 8Δ = 0, the energy of one external DoF that can be experimen- tally detected at the GT. The experimental value of IE energy for flexible sys- tem is Δ 0 ( i) = Δ ≈ 5.6 104 eV from WLF equation [18]; and Δ ≈ 5.5 104 eV from ΔEco on melt spin- ning-line [8,10]. So 0( i) = 8Δ ≈ 4.4 103 eV. This value is consistent with the experimental results (4 12 103 eV) for Boson peak measured by high-resolution inelastic neutron scattering [12]. For non-flexible general Figure 2. Two-body delocalization along 8 orders of geodesic, the fixed point of 15/16 0( i) is the universal constant for all self-similar molecule-clusters i with inverse cascade–cascade mode in random systems.  J.-L. Wu / Natural Science 3 (2011) 580-593 Copyright © 2011 SciRes. OPEN ACCESS 583 system, each of 320 IEs may have different IE energy. Thus, each material has its own two inherent values of independent of temperature in its own random system: the geometric frustration—percolation (also the localiza- tion—delocalization) transition energy, Ec = kTg° (vi), and the renewed cluster energy, kTm°(vi). kTg°(vi) is also the non-integrable directional regression vibration (DRV) kinetic energy of i-th order cluster vi, kTg°(v8) = kTg in inverse cascade; and kTm°(vi) = kTg°(vi) + 4 0(vi), kTm°(v8) = kTm, of the 8 orders of clusters in cascade [10]. 2.2. Origin of Interface Excitation in Statistical Physics This theory differs from current mode-coupling theo- ries [11] of GT. A phenomenological concept of IE is introduced by the ripplon and the universal Lindemann distance increment [10] dL in order to obtain the global properties of IEs in inverse cascade – cascade. The spirit in mode-coupling scheme deals with the coupling of fast-slow relaxation modes and two density modes in structure rearrangements at GT. In our mode-coupling scheme, we first focus on finding out the three direction non-integrable energies kT2°, kTg° and kTm° existing in the coordinates invalidation from i-th order clusters to (i + 1)-th order at GT, in spite of the time complications in all anharmonic frequencies. This means that the crucial 2D 2 closed loop with the maximum non-integrable Berry’s Phase plays a major role in anharmonic fre- quency. The mode-coupling trick is that the relaxation time complications have been beforehand reduced and replaced by the slow Brownian directional regressions. Then we deduce from global to local to find out the mi- croscopic origin of IE (Section 2.3). The mosaic struc- ture theory of GT reaches a new level by revealing the microscopic physical origin of IE. The fluctuation stability condition in (ii) is that the chemical potentials are always zero in all subsystems [15]. There is a profound theoretical contrast between chemical potential and ‘temperature’. The chemical po- tential is defined as Fermi energy level on the occasion when the random kinetic energy of temperature in sys- tem is zero in solid physics; whereas the non-integrable random DRV energy, kTg°, in a thaw domain, is defined as the delocalization energy, Ec, on the occasion when the chemical potentials in all subsystems are always zero at the solid-to-liquid GT. The key here is that the kTg is only comprised by the slowest (z-space) DRV energy, which is exactly the lowest-frequency ( i scale) mode that has been explored at the GT. The x-axial and y-axial ( i scale) DRV energy of all z-component molecules (chain-particles) encircled by IEs should be zero in order to minimize the total dominant GT energy. In Figure 2, there are 8 sharp-angled points, U c* = U(qi,R) = U(qi+1, L) [14], forming the delocalization paths of two z-axial (i.e. q-axial in the figure) 1 clusters along 8 orders of geodesic with IE energy (τi) = 1/8 0(τi). i-th order geodesic is the shortest line of 2 cycle be- tween qi,R and qi+1,R on i-th order of cylindric potential surface (on x-y projection plane, taking 0 = 1 as z-coordinate axis). The red arrows denote the paths of structure rearrangements in cascade: from q8,R → q7,R → …→ q1,R → q0,R. 2.3. Absence of Attraction in 2D Lattices Thus, two 1 clusters are in the delocalization state, absent of attraction in vibration, along 8 orders of geo- desic. The two molecule-clusters in the absence of Van der Waals attraction are all through in a repulsion state on the x-y projection plane during generating their one IE. Van der Waals interaction includes the contribution of instantaneous induced dipole - induced dipole. Generally, instantaneous polarized dipole electron ch- arges randomly distribute on interface 1-2 forming elec- tron cloud (blue zone) in Figure 3(a). At GT, an interesting and unexplored corner in Van der Waals interaction theories is that the synchronal in- stantaneous polarized electron charge coupling pair (two small blue dots) may parallel transport on an interface 1-2 between chain-particles a0 and b0, or the interface 2-3 between a0 and c0 to form IE in Figure 3(b), or 3(c). Since the site-phase difference on x-y plane between the two z-component molecules is [10], the state of the all polarized electron charges in each instantaneous dipole, in the two z-component molecules in Figure 3(b-1) or 3(c-1), must be in the same state and in the z-axial min- imum energy state (ground state) of single-molecule in- stantaneous dipole. In Figure 3, (b-2) and (c-2) is re- spectively the projection of (b-1) and (c-1) on x-y plane. Thus, the additional IE energy is the repulsive energy between the two instantaneous synchrony z-axial ground state polarized electron charges that parallel transport, from one end to other end on an interface in Figure 3(b-2), simply denoted by an arrowhead (12) in 3(d) on local x-y projection plane. Note that the two IE states of (b-1) and (c-1) in Figure 3 occur at different local instantaneous times. The directions of the next two par- allel transports of instantaneous polarized electron charge of the reference a0 are denoted as two red-broken line arrowheads in Figure 3(c-1). IE loop-flow 1 2 3 4 1 in Figure 3(d) offers a non-integrable poten- tial to induce ion a0 (the center of mass of a0 particle) z-direction a displacement z. Black-broken line arrow denotes the delocalizing direction of each ion in Figure 3. Figure 3 explains the origin of 5-particle cooperative excited field in [10]. The molecules in the absence of the x-, y-axial ( i scale) vibrations are in the high density 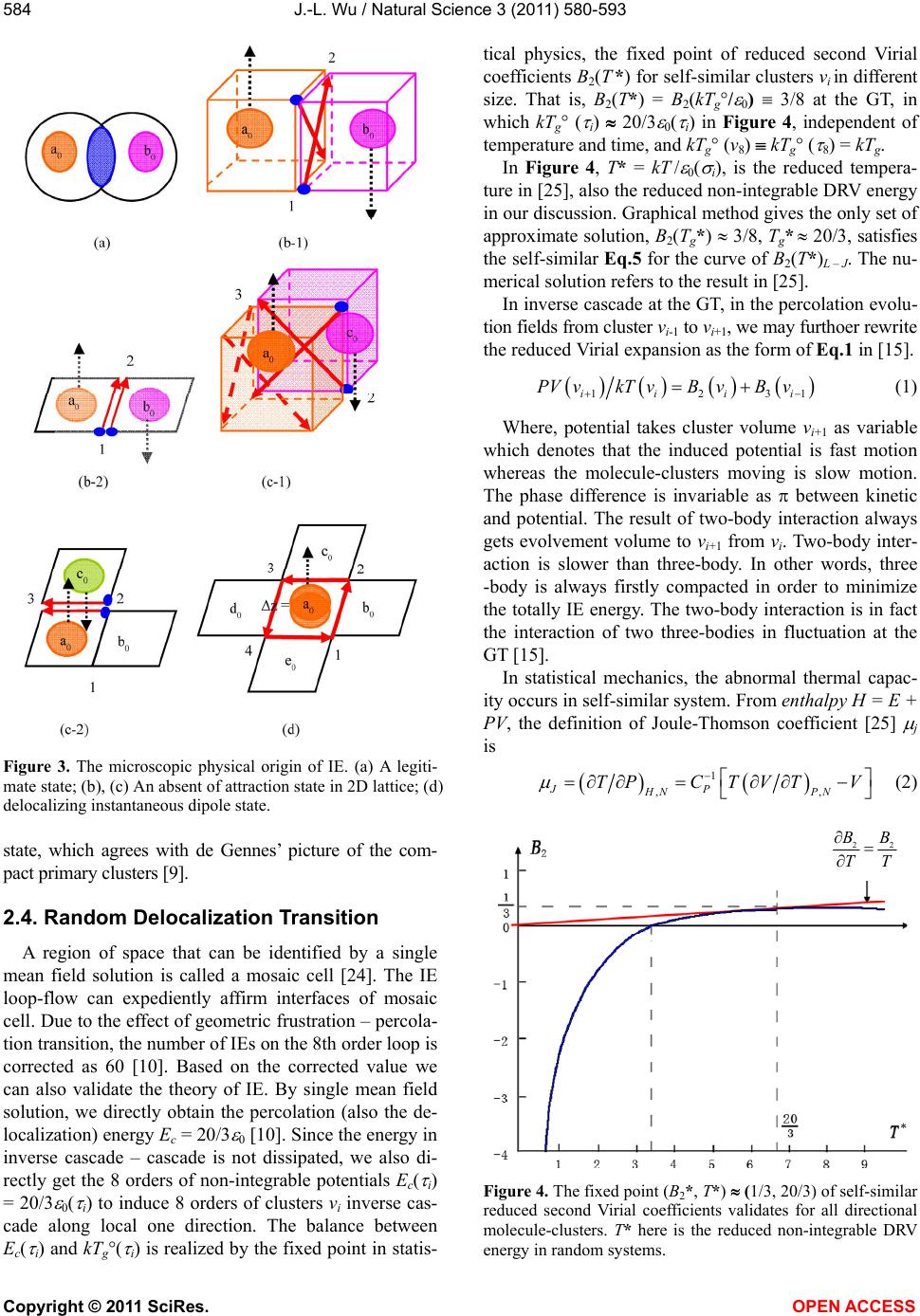 J.-L. Wu / Natural Science 3 (2011) 580-593 Copyright © 2011 SciRes. OPEN ACCESS 584 Figure 3. The microscopic physical origin of IE. (a) A legiti- mate state; (b), (c) An absent of attraction state in 2D lattice; (d) delocalizing instantaneous dipole state. state, which agrees with de Gennes’ picture of the com- pact primary clusters [9]. 2.4. Random Delocalization Transition A region of space that can be identified by a single mean field solution is called a mosaic cell [24]. The IE loop-flow can expediently affirm interfaces of mosaic cell. Due to the effect of geometric frustration – percola- tion transition, the number of IEs on the 8th order loop is corrected as 60 [10]. Based on the corrected value we can also validate the theory of IE. By single mean field solution, we directly obtain the percolation (also the de- localization) energy Ec = 20/3 0 [10]. Since the energy in inverse cascade – cascade is not dissipated, we also di- rectly get the 8 orders of non-integrable potentials Ec( i) = 20/3 0( i) to induce 8 orders of clusters vi inverse cas- cade along local one direction. The balance between Ec( i) and kTg( i) is realized by the fixed point in statis- tical physics, the fixed point of reduced second Virial coefficients B2(T *) for self-similar clusters vi in different size. That is, B2(T*) = B2(kTg°/ 0) 3/8 at the GT, in which kTg° ( i) 20/3 0( i) in Figure 4, independent of temperature and time, and kTg° (v8) kTg° ( 8) = kTg. In Figure 4, T* = kT / 0( i), is the reduced tempera- ture in [25], also the reduced non-integrable DRV energy in our discussion. Graphical method gives the only set of approximate solution, B2(Tg*) 3/8, Tg* 20/3, satisfies the self-similar Eq.5 for the curve of B2(T*)L – J. The nu- merical solution refers to the result in [25]. In inverse cascade at the GT, in the percolation evolu- tion fields from cluster vi-1 to vi+1, we may furthoer rewrite the reduced Virial expansion as the form of Eq.1 in [15]. 1231iiii PV vkT vBvBv (1) Where, potential takes cluster volume vi+1 as variable which denotes that the induced potential is fast motion whereas the molecule-clusters moving is slow motion. The phase difference is invariable as between kinetic and potential. The result of two-body interaction always gets evolvement volume to vi+1 from vi. Two-body inter- action is slower than three-body. In other words, three -body is always firstly compacted in order to minimize the totally IE energy. The two-body interaction is in fact the interaction of two three-bodies in fluctuation at the GT [15]. In statistical mechanics, the abnormal thermal capac- ity occurs in self-similar system. From enthalpy H = E + PV, the definition of Joule-Thomson coefficient [25] j is 1 ,, JP HN PN TPCT VTV (2) 22 BB TT Figure 4. The fixed point (B2*, T*) (1/3, 20/3) of self-similar reduced second Virial coefficients validates for all directional molecule-clusters. T* here is the reduced non-integrable DRV energy in random systems.  J.-L. Wu / Natural Science 3 (2011) 580-593 Copyright © 2011 SciRes. OPEN ACCESS 585 Assume the state of J 0 corresponds to GT. When J 0 ,PN VT VT (3) Rewrite as Eq.4 for vi cluster , ii ii PN Vv kTvVv kTv (4) Here kT in (2) (4) should be also regarded as the non-integrable random DRV energy of i-th order cluster vi in z-space. The energy kT also is a function of the self-similar cluster volume v i, when outside pressure (stress) remains constant. As long as the condition J 0 in (2) is satisfied and (4) also holds, CP in the case of (4) may also show an abnormal change. We see the abno r- mal thermal capacity may occur in the following two cases. One is in the polymer GT, CP occurs at the tem- perature Tg. In this case, the kTg (= Ec) is the percolation energy. The other is in the low-temperature GT in local domain region, the abnormal thermal capacity occurs in the manner of the energy of Boson Perk. The hard-sphere (square well potential with IE energy) model at the GT can be also deduced from the 8 orders of self-similar L-J potentials along the clusters inverse cascade direction [14]. It can be strictly proved that the reduced third Virial coefficient for hard-sphere system is constant, B3 5/8, [25,26] independent of temperature and cluster volume. Thus, in z-space of the clusters in- verse cascade, we can also get B3(vi-1) 5/8. Therefore, in the percolation evolving field from cluster v i-1 to v i, from the interaction of two three-bodies, we have 2ii i PV vkT vBv in z-space fluctuation. In z-space, from (1), 22ii iii Bv Bv P Vv kTv Vv From (4), we obtain an important self-similar Eq.5 at the GT in [15]. 22ii ii Bv Bv kTv kTv (5) It Figure 4, if we regard the energy of T g* as the z-space reduced DRV kinetic energy of 8 orders of self-similar clusters, the point (B2, kTg°) (3/8, 20/3 0) is exactly the universal geometric frustration percola- tion transition energy for any cluster volume v in random delocalization transitions. Moreover, the B2 3/8 at the GT is directly proved by scaling theoretical approach [15]. 1 23-1 3858 1 i ii i PV vBv Bv kT v (6) where i = 1, 2,…, 7, and when i = 8, the induced poten- tial Ec directly equals to kTg in the geometric frustration – percolation transition at GT. Eq.6 holds true on all subsystems (flow-percolation fields), which means that the kinetic energy always keeps balances with the poten- tial energy, in the manner of the maximum Berry’s phase of , in the mode of 8 orders of coupling 2body-3body fluctuation clusters at GT. 2.5. Order-Induced Molecule Delocalization The concept of connecting Anderson transition and GT is the percolation limit model. In the theory of An- derson disorder-induced (electron) localization [13], the competition between kinetic energy and potential energy influences on the electron states can reside in the ratio W/B, W, the magnitude of the random potential, and B, the (crystal) bandwidth in the absence of disorder. A classical method of demonstrating consequences of dis- order is the percolation model [13]. At the GT, the per- colation transition energy is Ec( 8) = Ec. ΔEco is the ori- entation activation energy with 320 IEs on the 8-order 2D mosaic structure in local z-space [10]. The ratio of Ec /ΔEco c(Ec) 1/6, its physical meaning is that c(Ec) specifies the occupied fraction of z-space 320 IE states that allow flow of energy Ec to occupy. The reason of the cooperative molecules obtaining the probability of delo- calization is that they share energy Ec. The ratio is con- sistent with the result (the classical analog for electron delocalization) of Zallen [13], who suggests c (Ec) 0.16 1/6, for the percolation limit on a continuum in 3D space. Here, the occupied fraction of all IE states allowed to flow of energy Ec, takes the place of the oc- cupied fraction of space allowed to molecules of energy Ec. This means that there is the inherent theoretical con- trast between molecule delocalization and electron lo- calization on the concept of percolation limit. That is, from the viewpoint of percolation, there are two classical percolation limits. One is the limit of the maximum disord er for the potential in random corre- sponds to the Anderson transition, that is the Zallen’s viewpoint [13]; whereas, the other is the limit of the maximum order for the potential in random to the GT. At the GT, all molecules encircled by 4 IEs on x-y projec- tion plane are in the minimum excited energy state (states), i.e., at the z-axial ground state of single - mole- cule dipole in the absence of x-, y-axial ( i scale) vibra- tions. The DRV induced potential energy in 5-particle cooperative excited field [10] can be also regarded as the overall order potential limit (in contrast to the maximum disorder potential limit in Anderson random) in the ran- dom Brownian motion. That is the reason we call the GT as the order-induced molecule (molecule-cluster) delo- calization transition in random systems. In other words, the delocalization energy Ec at the GT  J.-L. Wu / Natural Science 3 (2011) 580-593 Copyright © 2011 SciRes. OPEN ACCESS 586 comes from the maximum order energy in random and the minimum additional ground-state repulsive energy. Thus, the percolation limit mode (the crossover from the site- or bond- percolation in solid to the flow-percolation in liquid) of the GT can probably explain why the 8 or- ders of 2D mosaic geometric structure (Figure 1) asso- ciated with percolation transition so simple. That is, this nano-scale soft mosaic structure reflects the nature of random delocalization transition in random systems. 2.6. Percolation Limit Picture at GT The theory of IE loop-flow directly and definitely shows Emig = kT2 = 17/3 0. Emig is defined as the average energy of cooperative migration along one direction in a percolation field [10], independent of temperature. Dur- ing [ti, ti + ti] in a reference domain, the entire transient i scale displacement energy in z-axial is (136/8) 0 = 17 0, Figure 1, and the i scale vibration energy in x-, y-axial is zero; thus, the average DRV kinetic energy is also 17/3 0 = kT2. On the other hand, during (ti + ti, ti+1), the average random energy in the domain is as kT2. However, the average DRV kinetic energy would occur in other domains in the system during (ti + ti, ti+1) in the reference domain at the GT. This is the dual-role of kT2 in kTg, reflecting the balance between random DRV en- ergy and random thermal motion energy at the condition of kTg°. If take Emig = kT2 as the average DRV kinetic energy in dual-role, kT2 is in fact the deferred action DRV kinetic energy until the appearance of the 8th order 2D loop (the appearance of kTg). In this case, kT2 will be of the DRV kinetic energy of the 8th order cluster in the absent of energy 0( 8) in an excited domain in Figure 1. Vice versa, when the temperature is T2, the reference domain will also be of the probability of regression order potential of kT2 if the domain can obtain the energy 0( 8). Thus, the physical meaning of kT2 is that in a 3D solid-domain, taking any reference direction, as long as superadded the energy 0( 8) on the domain, the sponta- neous molecules delocalization would be occur in the reference direction. Its singularity different from phase transition is the non-ergodicity that the adding energy may be either a few energies only on several domains, or a large energies one by one on neighboring domains to form a flow- -percolation field. In any case, if the adding energy 0 denoted by temperature, it always satisfies kT2 + 0 = kTg. In other words, here 0 = 0( 8) is a reduced energy to reduce the magnitude of its action region or its action time in the system. The noticeable contrast between phase transition and random transition in physical the- ory (Table 1) is as follow. At Curie temperature point, kTc, in magnetism, so as decrease a little of rando m en- ergy (kT) in whole system, the spontaneous magnetiza- tion will occur along one direction in whole system. Whereas, at kT2 of the GT, so as increas e a little of di- rectional ordering energy 0 in a local region, the mole- cules spontaneous delocalization will occur along the direction in the local region. It is in this sense T2 corre- sponds to Curie temperature in magnetism. The numeri- cal value of 17/3 0 is also validated by the WLF experi- mental equation [18], 17/3 0 + 0 = kTg = 20/3 0 = Ec. There are two critical delocalization energies Ec and kT2. Ec is the critical percolation transition energy, corre- sponding to the spontaneous delocalized solitary wave (Section 3) in percolation field (the “ocean” in percola- tion theory) at GT; and kT2 is the critical energy of parti- cle-clusters delocalization at low-temperature GT in lo- cal domain scale (the “lake” in percolation theory) in percolation limit model. This percolation limit picture with separate ‘lakes’ and ‘ocean’ is distinctly different from the general percolation model. Note: the delocaliz- ing step-size in glassy is far less than the amplitude of thermal vibration, Section 3.3, thus the glass state in random delocalizing is still in stable state. 2.7. Soft Matrix and Emergent Property The de Gennes’ central assumption of the soft matrix [9] is also validated. The soft matrix is exactly i-th (i 7) order 2D loop-flows frozen in glass state. From which the low-temperature GT would occur through a longer Brownian regression time to inverse cascade until the 8th order and to cascade at low-temperature. The lower the frozen, the longer is the required time of inverse cascade. Therefore, the abnormal exponential function [27] in glass state can be proved directly. Generally, the physical quantity (t) in a system will return according to the physical law of Eq.7 when the system deviates from its equilibrium state. ~exp t t (7) However, if the glassy system is driven (or normally fluctuates) out of equilibrium, it returns according to the formula [28] exp t Where t is the (system) time and and are parame- ters. Unfortunately this is not a mathematical expression that is frequently encountered in physics. So little idea exists of what the underlying mechanisms are [28]. Since the inverse cascade–cascade motions only occur in some discrete “lakes” in glass state when T Tg. Eq.7 still holds true in these “lakes” regions as long as the t in (7) is the local domain time. One of the key concepts is  J.-L. Wu / Natural Science 3 (2011) 580-593 Copyright © 2011 SciRes. OPEN ACCESS 587 that the equilibrium state of glass state is the equilibrium state between the random thermo motion energy kT and the slowest DRV energy. DRV energy always dominates the number of the excited domains at temperature T. Assume the glass state we observed is the nonequilib- rium state that comes from the equilibrium state at the temperature T1 suddenly drops to the temperature T2 at the time t = 0, and T2 T1 Tg. During the relaxation time of t, the entire DRV relaxation energy is = k(T1 T2) = kT. From the famous Kolmogorov law in cascade [1,29] 2 3constant i i i kTt l t . (8) Where li is the length scale of i-th order of loop (clus- ter) and i is the cascade energy mobility; ti is the local domain time and t is the relaxation time in system. From (8) 1 23 i i tl kT t (9) Substituting ti in (9) for t of local domain time in the right term on (7) 1 23 ~exp exp s loc i ys tl kT tt (10) The physical meaning of is very definitude, =1/3 denotes the cascade motion in ideal glass state in the case of the small fluctuation value of kT. The reason of the deviation from 1/3 for may be that (a) the influence of the competition between the change ratio of tempera- ture dT/dt and the 8 orders of relaxation times on IEs; (b) the larger value of kT conduces augment because of the inverse cascade always lower than cascade. Similarly, if the nonequilibrium glass state is arose by outside stress (or electromagnetism and other factors) work W, the relaxation of W can only realize through the inverse cascade – cascade in the excited domains as same as that in the proof of WLF equation [18]. In this case, we only need replace kT by W in (9). This means that the ab- normal mathematical expression (10) in glass state is also only the emergent property of domains in system, as same as the abnormal expression of WLF equation in the GT (the many-times repeated applications of Clape- yron equation governing first order phase transition on the subsystems will educe WLF equation) [18]. The general physical relaxation law (7) still holds true in domain scale in glass state. The theoretical proof also confirms that inverse cascade–cascade mode is the fun- damental mode for glass state. 3. ENTANGLEMENT STRUCTURE 3.1. Theoretical Proof of the 3.4 Power Law The viscosity in ME melt is N3.4 for all linear en- tangled polymers. Since N is a large number, the ex- perimental 3.4 power law of viscosity has sensitivity for any modified theory to de Gennes reptation model. In concordance with the experimental 3.4 power law of viscosity, within the experimental error range of viscos- ity, we first roughly estimate the theoretical error toler- ance by adopting a fine theory described ME viscosity. The critical entanglement chain length Nc = 200 [11,30]. The experimental value of chain length N is generally less than 103 (seeing the Fig. 9.5 in [5]). If taking the range of N as 200 1000, N3.4 /N3.3 1.7 (N = 200) 2 (N = 1000). This indicates the error between theoretical value and experimental result of viscosity will be about 70% 100% if the theoretical value as 3.3; and N3.4/ N3.35 1.3 1.4. This means that a fine theory should be able to give the theoretical exponential value range of viscosity as 3.4 0.05 for flexible polymers. At the same time, we expect this theory must also be able to predict the exponential of viscosity that is in line with the ex- perimental results for non-flexible polymers. This is also a fine way to check up the GT theory. The existing vari- ous modified theories [5] cannot match for the 3.4 power law. This exponent is significantly large than the predic- tion of 3 by de Gennes reptation model [5,30] based on the assumption that the chain of length N is a “free chain”. The reason of the deviation is the required num- ber of DoF of one-step-walk along z-axial for each chain particle depends on Nz the z-component of chain N. The key of the theoretical proof is to find out the number of DoF, N*, for chain-length N. Only through substitution of the reference chain of length N in single-chain reptation model with an equivalent particle-chain of length N*, can the random diffuse motion of the reference chain N be entirely free in tube model. Thus, in the ‘equivalent mul- tichain’ de Gennes reptation model, we have 3 ~N (11) The one-step-walk along +z-axial of a reference parti- cle a0 on chain Nz results from the induced action of the 8 orders of 2D loop-flows on the x-y projection plane in a0 (local excited) field. Only when the 8th order appears and acts on the particle a0, should one “particle-cavity” in +z-direction appear in a0 field and bring a0 to move one step along +z-axial. Statistically, when Nz Nc = 200 (Nc is also the number of the chain-particles of structure rearrangements [10,11,30,31] in the 8 orders of 2D mo- saic structure), the moving of the 200 cooperative parti- cles along z-space on x-y projection plane in a0 field  J.-L. Wu / Natural Science 3 (2011) 580-593 Copyright © 2011 SciRes. OPEN ACCESS 588 should share the energy Ec with a0. Due to the sharing energy effect, even if a0 has obtained a cavity with one-step-walk, the motion of a0 should still be correlated with the 200 particles (molecules) in shared energy Ec. In other words, the cooperative particles still “drag” the one-step-walk of a0. The external DoF of the 8th order 2D loop-flow en- circled a0 is 1, taking 0( 8) as the energy unit of DoF. In order to eliminate the correlation of share energy to make a0 move freely in z-axial, let Ec / 0( 8) = Lg, where Lg is the equivalent number of particles. Ec thus is as the “loop-flow” energy with an equivalent chain of length Lg circling a0. On the one hand, from the viewpoint of par- ticle a0 on chain Nz, statistically, the Nc chain-particles on Nz also share the energy Ec. Thus, the probability that a0 is possessed of one unit DoF on the chain Nz is 1/N. On the other hand, from the viewpoint of the Lg equiva- lent particles, statistically, each equivalent particle is located at its own long-chain with length Nz, and the probability that each equivalent particle is possessed of one unit DoF on its own long-chain is also 1/N. When particle a0 on chain Nz is substituted by an equivalent chain of length Lg, the probability of the event that it obtains one unit DoF and freely moves one step along z-axial, denoted as p+(a0). p+(a0) is namely the probability of the event that all the Lg equivalent parti- cles simultaneity move one step along z-direction in the co-Brownian motion by 200Nz chain-particles in a0 field. So, p+(a0) is given as 01 pa N (12) Statistically, the probability of each particle freely moving one step in z-space should be all equal to p+(a0). To maintain the balance of motion between inverse cas- cade and cascade in z-space, the probability that one reference 8th 2D loop-flow disappears in cascade in co-Brownian motion by 200Nz chain-particles is denoted as p(a0) and obtained as follows. Let a0 have nz DoFs with 0( 8) as the unit in cascade motion. nz is actually the number of DoF to make con- formational rearrangement of the chain Nz in z-space, which results from the contribution of the reference 8th order 2D loop-flow transferred from the particle a0 field to the other particle field on the chain. Similar to (12), we have 0 m z pan (13) where Lm = kTm / 0( 8), Lm is the number of ‘equivalent particles’ to eliminate the reference 8th order 2D loop-flow of a0 in cascade. In the complex inverse cascade–cascade motion, the equilibrium condition is 00 pa pap (14) so, 1 m T T z nN (15) The number of DoF of Nz chain-particles on chain of length Nz to make conformational rearrangement in z-space is Nz *, Nz * = nz Nz, 1 * m T T z NN (16) In (16), Tg° has been approximately replaced by Tg. Nz * 0( 8) H, here H is enthalpy in (2), the constant en- ergy for rearrange conformation (that also the energy of z-space solitary wave in Section 3.3) in random system. So, during the reptation of long-chain N in the 3D space, the number of DoF Nx * in the x-space and Ny * in the y-space have Nz * = Ny * = Nx *, or 31 * m T T NN (17) From (11) and (17), we obtain the general power law expression of viscosity in ME 91 m T T N (18) For flexible chain polymers, kTm = kTg + 4 0( 8), kTg = 20/3 0( 8) [10]. We have 9 8 * NN (19) And 27 83.375 3.4 ηNN N This theoretical result conforms well with the experi- mental data. For non-flexible chain polymer, Eq.18 can be verified. For example, for the polypropylene (PP), the Tg(PP) is 10C = 263k [32]; the Tm(PP) is 176C = 449k [33]; Eq.18 given pp(theory) N3.73, conforms well with the experimental data pp(experiment) N3.72 [34]. The theoretical proof further confirms that inverse cascade–cascade mode is the fundamental mode in the solid-to-liquid transition whether in macromolecular or in small molecular. 3.2. Degrees of Freedom of Chain-Length N Since cascade is also the mode in turbulent flow, we discuss the total number of DoF, NL, in the cube with edge length of L (L N*) in ME melts. The unit for the length of L here is the loop scale, ls, of the 8th order 2D loop-flow. Due to the properties of flow-percolation [10], an 8th order 2D loop-flow may be in arbitrary direction. In other words, in 3D space, we can consider any one  J.-L. Wu / Natural Science 3 (2011) 580-593 Copyright © 2011 SciRes. OPEN ACCESS 589 “random-walk chain” with “chain-length” NL and each “chain-particle” with one unit length l s. If the “end-to- -end distance” of the “random-walk chain” is always equal to L, L is then the root-mean-square of end-to-end “chain-length” NL, that is, NL = L2, in Figure 5. When L N*, NL (N*)2, from (19), we have 94 ~ L NN (20) Comparing to the famous Kolmogorov relationship [29] between the number of DoF NR and the Reynolds numbers Re in (a uniform grid) turbulence 94 ~ Re NR (21) It can be seen that the two terms in two different sub- jects: turbulence and ME both describe the same com- plex motion phenomena about inverse cascade – cascade. Both of them obey the same scaling law, which indicates there is a universal theory behind them. Reynolds num- ber is actually the ratio of the inertia force to the drag force, and the macromolecular chain-length reflects the ratio of the number of the particles “inertially” one by one sharing the delocalization energy Ec to the one parti- cle entangled with them in motion. 3.3. Solitary Wave Each physical quantity in (12) (18) is the inherent invariable in random system for every material, inde- pendent of temperature and time. Eqs.15 and 16 reveal a z-space solitary wave moving N z steps along a random long-chain Nz from one end to other end. A distinct cha- racteristic of the solitary wave is that the “particle” en- ergy of each one-step-walk is “quantized”: nz 0( i) = nz 0 for flexible system. While nz 0( i) generally have 8 or- ders of components, 8 orders of potential well energies for general non-flexible system. The step-size is only the nz particles of size. The number of step of its traveling wave is Nz = N. The reptation in 3D space of a ME chain is assembled in time by the three solitary waves on the chain in x-, y- and z-space, which in fact attributes to the co-Brownian regression motion by 200N chain-particles respectively in x-, y- and z-space. Accordingly, the ME structure about viscosity is in fact the 8 orders of transient 2D mosaic structures on x-y projection plane and the delocalized solitary wave in z-space. nz is a small numerical value, e.g. if taking N = Nc = 200, nz 0.036. However, Eq.15 reveals that nz connects with the length-boundaries Nz of solitary wave in its each one-step-walk. The velocity of traveling wave will be determined by other factors including, e.g., the generating ratio of IEs by temperature, time or outside stress work, and the magnitudes of material relaxation times in IEs. This special characteristic of solitary wave shows that it differs from the mode of conventional quan- Figure 5. The relationship between “chain-length” NL and number of degrees of freedom. tum-mechanical wave; the latter is always obtained from the combination of both the wave equation and the bou- ndary conditions. 3.4. Spontaneous Symmetry Breaking at GT The paradigm for the phase transition theory is the ferromagnetic transition. de Gennes [30] emphasized that polymer solutions should compare with magnets. However, difficulty occurs in the order parameter for a polymer solution [30]. de Gennes proposed that the or- der parameter is the magnetization of a spin system with a number of spin components n = 0 [30]. Edwards’ more concrete statement is that the order parameter is simi- lar to a quantum mechanical creation (or destruction) operator [30]. The proof for 3.4 power law predicts a definitude result for the remaining problem that the order parameter is the spontaneous delocalized solitary wave of a classical spin system (8 orders of 2D IE loops) with a number of additional instantaneous spin compo- nents n = 0, whether in macromolecules or in small molecules, within the entire range from solid to liquid transition. The physical meaning of n = 0 is here that the additional “spin” of a reference chain-particle only comes from the contributions of its 4 neighbouring par- ticle fields, instead of its own inner-rotation change. In addition, n = 0 also corresponding to self-avoiding ran- dom walk [30] of 8th order 2D loop-flows. Contrary to phase transition, we have unveiled the step by step way from disorder to more disorder in Table 1. In which we have implied that the disordered degree (measurement) depends on the maximum 2D IE loop-size and the number of the loops in a random sys- tem. The conjugate variables of solid-to-liquid transition in Table 1 are deduced from the results of de Gennes in [30].  J.-L. Wu / Natural Science 3 (2011) 580-593 Copyright © 2011 SciRes. OPEN ACCESS 590 Table 1. The contrast between delocalization transition and phase transition in 3D space. Critical point Ferromagnetic phase transition Solid-to-liquid delocalization transition Number of spin components n n = 3 n = 0 Cell Magnetic moment S displacement energy circled by 4 IEs Spontaneity Magnetization M Delocalizing solitary wave “Curie” point Tc kT2: critical delocalization energy Reduced energy increment = k (T Tc) / kTc , 0, M = 0 in system 0, M = S 0 in system = k(T T2) / 0, 0: potential well energy = 1, GT: 0 in subsystems 1, Low-temperate GT: 0 in domain = 5, MT: Reptation in subsystems 1 5, Viscoelasticity (Intermittency) Symmetry breaking M chooses one-space, breaking the isotropic M GT: Solitary wave chooses one space, breaking the isotropic thermal motion MT: Three solitary waves in x, y, z-space restore the isotropic thermal motion Self-similar cluster cluster size v Finite 8th order cluster Correlation length =Nz, macromolecular chain-length =Nc, for small molecule systems Order parameter M Solitary wave Replica symmetry Spin wave 8 orders of 2D IE loop-flows Conjugate variables Extensive quantity Intensive quantity M H Nz (or Nc), step number of solitary wave nz 0, one-step-energy of solitary wave Generalized rigidity Magnetic hysteresis 8th order 2D IE loop Defect Domain boundary Extra vacancy volume in each IE on 8th order loop 4. COHERENT STRUCTURE 4.1. The Way to Turbulence Thus, we have ascribed the GT problem to the syste- matics of the special parallel transport state of bonding on intermolecular interface. This is precisely in line with the insight in [28] that crucial to the endeavor of GT theory is a deeper understanding of the systematics of bonding in condensed matter within a framework going considerable beyond the current GT picture. All that we have done is to replace the GT problem by the bonding problem of parallel transport of instantaneous polarized electron charges containing all atoms (atom-clusters) in a chain-particle at the GT. However, we have progressed and can now discuss turbulence. The IE and the 8 orders of 2D mosaic structures at GT may be called as the ground state IE and the first level of 8 orders of 2D mosaic structures. For small molecule system, due to the energy transfer of rearrangement conformation between two z-axial connecting molecules, Nc z-component molecules respectively in absence of x-, y-axial vibrations can also be connected one by one to form a z-space random “long-chain with chain-section as a molecule” walking Nc steps in the correlation of shar- ing energy Ec. Solid-to-liquid transition does not invoke the energy level of molecule (molecule-cluster) instantaneous in- duced dipole. However, for each energy level of mole- cule (molecule-cluster) instantaneous polarized dipole, e.g., for the first energy level of instantaneous polarized electron charges of molecule (of all atoms and molecules in a molecule-cluster), two neighboring molecules with antiparallel delocalization may be all in the z-axial first energy level state of single-molecule instantaneous po- larized electron charges on an intermolecular interface. Such IE is named as the first-energy-level IE. A new 8 orders of 2D mosaic structure can be formed, which is called the second level of 8 orders of 2D coherent (mo- saic) structure. For small molecule liquid, only about 4/15 (8kTm / Eco = 4/15) interfaces have been excited. As disorder increases, more ground state IE will be excited to accel- erate liquid flow, which corresponds to the ordinary flow development. As disorder keeps increasing and flow continues speedup, each ground state IE on the 8th order 2D loop can update to be the first-energy-level IE with new 8 orders of relaxation times, new additional IE energy and vacancy volume. All other IEs on x-y projection plane are still in the ground state of IE. All these z-component molecules in the absence of x-, y-axial vibrations can form a z-space random “long-chain” with “chain-section” as 136 molecules encircled by 60 first-energy-level IEs.  J.-L. Wu / Natural Science 3 (2011) 580-593 Copyright © 2011 SciRes. OPEN ACCESS 591 60 first-energy-level IEs can form a 0-th “cluster-par- ticle” in z-space in the second level of 8 orders of 2D loop-flow, denoted as v0 (1) (a0), the figure in the super- script parentheses denotes the first-energy-level IE of single-molecule polarized electron charges. The 0-th “cluster-particle” is self-similar to the z-component mo- lecule a0 [named as v0(a0)] in Figure 1. The size of the 0-th “cluster-particle” is that of the 8th order cluster v8(a0) in the first level of mosaic structures. The 0-th “cluster-particle” has also 4 “interfaces” with the side length of 15 IEs (one thick-black inverted arrow plus the 14 sky-blue arrows in Figure 1) of the first energy level. Thus, the second level of 8 orders of 2D mosaic struc- tures is self-similar to the first level. To drive the 136 molecules cooperatively move one step along +z-space only needs the energy nz (1) 0 (1). The mode is the solitary wave with the first-energy-level IE in z-space. 4.2. Multilevel of Random Localization - Delocalization Transitions Inverse cascade is the accumulation of the directional Brownian regression potential energy in loop-flows, and cascade is the transfer of energy from potential to kinetic to drive liquid cluster flow. The larger loop is formed in inverse cascade, the bigger liquid cluster is driven in cascade, and the higher efficiency the system has to ex- cite liquid flowing, and the faster the velocity of flow is. In the same way, when the flow keeps on speedup, the third, the forth…the l-th level of 8 orders of 2D coherent structures can be constructed one by one, in order to augment the loop scale and the loop-potential so that the maximu m 2D loop-potential reaches the value of the potential forced by environment to the liquid flow. At the moment, the l-th level geometric frustration – percola- tion transition (seeing the universal pictures of Figures 1 and 2) appears and reaches potential balances with ki- netic, and the cascade phenomenon of the maximum loop-flow begins. This is the turbulent flow. The origin of the intermittency turbulence [35] is that the high non-ergodic state of flowing arises suddenly to make the reduced energy increment, (l), of each level with a value slightly more than 1, similar to the viscoelasticity in Ta- ble 1. The heteroclinic orbits [36] in turbulence may be the flow-lattice edges of the l-th level 2D coherent struc- tures. The complicated turbulence can come down to the l-th level solitary wave in z-space and the l 1 reptations (snake-walks) of l 1 various levels (self-similar sub- systems) simultaneously occurring in 3D space. At critical phase transition, the self-similar cluster size is v , while it is only Nc in GT, Table 1, which is also the localization-delocalization scale of the first level in universal random delocalization transitions. In other words, on the way from disorder to more disorder, the cluster size of v in critical phase transition will be here divided up by l levels, Nc (l), corresponding to the cooperative localization – delocalization scale of l-th level, i.e. it is also divided into l levels of geometric fru- stration—percolation transitions, similar to Figures 1-5, in random localization—delocalization transitions on the way from disorder to more disorder. This is the way to turbulence. 5. CONCLUSIONS A theoretical perspective on the mosaic structure the- ory of GT has been proposed. The theoretical approach of GT connects closely the origin of turbulence. A new and central concept is the instantaneous parallel trans- port repulsive state absent of attraction in 2D lattices in current Van der Waals interaction theories. The solid to liquid GT is the paradigm of the inverse cascade – cas- cade mode and the first level of the way to disorder and turbulence. Nine physical ingredients, random, self- similar, two-body interaction, fluctuation, frustration, percolation, delocalization, Berry’s phase (parallel trans- port) and Brownian regression potential, have been in- corporated on IE loop-flows at the GT and the universal random delocalization transition. The theoretical proofs for the abnormal exponential function in glass state and the 3.4 power law of viscosity confirm that the inverse cascade–cascade mode is the fundamental mode whether in solid to liquid transition or in macromolecular motion. Due to the effect of incorpo- ration of physical ingredients, the different terms in dif- ferent subjects have been also incorporated. The inverse cascade–cascade mode with mosaic structure reflects the confluence of both the kinetic dimensions and the ther- modynamics at the GT. It can be also denoted by both the delocalization solitary wave (that is also the melt mode, the conformational rearrangement mode, structure rearrangement mode) along a local one-dimensional di- rection and the 8 orders of 2D IE loop-flows (its scale or size is also a domain scale, cage scale, local scale, cor- relation scale of two-body interaction, random localiza- tion-delocalization transition scale, maximum step-scale of random walk, geometric frustration scale and critical percolation scale in random systems) on the local pro- jection plane. Three different structure terms, the mosaic structure in GT, the entanglement structure in ME and the coherent structure in turbulence, are all in fact the transient 2D geometric structure formed by IE loop-flows in their own random system. The most important physical quan- tity correlating GT, ME and turbulence is the directional ordering induced potential of a maximum 2D IE loop- flow in random system. Once the loop-flow is formed,  J.-L. Wu / Natural Science 3 (2011) 580-593 Copyright © 2011 SciRes. OPEN ACCESS 592 the GT appears, the critical ME phenomenon arises, and the cascade in turbulence begins. 6. ACKNOWLEDGEMENTS The author is grateful to all colleagues he had the pleasure to col- laborate and interact, especially when he found the fundamental phys- ics origin for the orientation activation energy obtained experimentally on melt high- speed spinning-line in 1986. In particular, the author would like to thank, in random order, Yuan Tseh Lee and Sheng Hsien Lin of Academia Sinica (Taiwan), Yun Huang of Beijing University, Da- -Cheng Wu of Sichuan University for useful discussions. Support from the Academia Sinica (Taiwan), and the State Key Lab of Chemi- cal Fibers and Polymer Materials, Donghua University (Shanghai) is gratefully acknowledged. REFERENCES [1] Lesieur, M., Yaglom, A. and David, F. (2002) The trends of turbulence. Springer, Berlin. [2] Anderson, P.W. (1995) Through the Glass Lightly. Scence, 267, 1615-1616. doi:10.1126/science.267.5204.1615-e [3] Berry, M.V. (1998) Foreword to “Global properties of simple quantum systems—Berry’s phase and others”. http://www.phy.bris.ac.uk. [4] Kadanoff, L.P. (1991) Complex Structures from Simple Systems. Physics Today, 44, 9. doi:10.1063/1.2810017 [5] Rubinstein, M. and Colby, R.H. (2003) Polymer physics. New York Publisher, Oxford. [6] Doi, M. and Edwards S.F. (1988) The theory of polymer dynamics. New York Publisher, Oxford. [7] Aleiner, I.L., Altshuler, B.L. and Shlyapnikov, G.V. (2010) A finite-temperature phase transition for disordered weakly interacting bosons in one dimension. Nature Physics, 6, 900-904. doi:10.1038/nphys1758 [8] Wu, J.-L., Guan, D. and Quian, B. (1986) The character- istic behaviour of the stretch-orientation zone during high-speed PET spinning. International Polymer Proces- sing, 1, 25-31. [9] Gennes, P.G. (2002) A simple picture for structural glasses. Comptes Rendus Physique, 3, 1263-1268. doi:10.1016/S1631-0705(02)01387-7 [10] Wu, J.-L. (2009) Unified model theory of the glass tran- sition: (Part 1) intrinsic 8 orders of 2D mosaic geometric structure. Journal of Materials Science and Engineering, 3, 69-83. [11] Lubchenko, V. and Wolynes, P.G. (2003) Origin of the boson peak and thermal conductivity plateau in low temperature glasses. Proceedings of the National Acad- emy of Sciences, 100, 1515-1518. doi:10.1073/pnas.252786999 [12] Greaves, G.N., Meneau, F., Majérus, O., Jones, D.G. and Taylor, J. (2005) Identifying vibrations that destabilize crystals and characterize the glassy state. Science, 308, 1299-1302. doi:10.1126/science.1109411 [13] Zallen, R. (1983) The physics of amorphous solids. Wi- ley, New York. [14] Wu, J.-L. (2009) Unified model theory of the glass tran- sition: (Part 2) fixed point of self-similar Lennard-Jones potentials in the glass transition. Journal of Materials Science and Engineering, 3, 58-64. [15] Wu, J.-L. (2009) Unified model theory of the glass tran- sition: (Part 3) fixed point of second virial coefficients in the glass transition. Journal of Materials Science and Engineering, 3, 31-39. [16] Laird, B.B. and Bembenek, S.D. (1996) Localization and the Glass Transition, Journal of Physics: Condensed Ma- tter, 8, Article ID: 9569. doi:10.1088/0953-8984/8/47/064 [17] Barkai, E., Naumov, E.A., Vainer, Yu.G., Bauer, M. and Kador, L.L. (2003) Statistics for random single-mo- lecule line shapes in a glass. Physical Review Letters, 91, 1-4. doi:10.1103/PhysRevLett.91.075502 [18] Wu, J.-L. (2009) Unified model theory of the glass tran- sition: (Part 4) Theoretical proof of the standard WLF equation in the glass transition. Journal of Materials Sci- ence and Engineering, 3, 76-82. [19] Mechler, S., Macht, M.-P., Schumacher, G., Zizak, I. and Wanderka, N. (2010) Frustration of the stable Zr-Ti-Ni quasicrystal as the basis of glass formation. Physical Re- view B, 81, 1-4. doi:10.1103/PhysRevB.81.180101 [20] Coslovich, D. and Pastore, G. (2007) Understanding fra- gility in supercooled Lennard-Jones mixtures: I. Locally preferred structures. Journal of Chemical Physics, 127, 1-13. doi:10.1063/1.2773716 [21] Shin, H.K. (2010) Vibrational relaxation of NO-(v = 1) in icosahedral (Ar) 12NO-clusters. Journal of Chemical Physics, 132, Article ID: 104302. doi:10.1063/1.3339385 [22] Tanaka, H. (2003) Roles of local icosahedral chemical ordering in glass and quasicrystal formation in metallic glass formers. Journal Physics: Condensed Matter, 15, 491-498. doi:10.1088/0953-8984/15/31/102 [23] Jain, T.S. and de Pablo, J.J. (2005) Local structure and motions in a supercooled polymer. Journal of Chemical Physics, 122, Article ID: 174515. doi:10.1063/1.1888505 [24] Lubchenko, V. and Wolynes, P.G. (2003) Barrier soften- ing near the onset of nonactivated transport in super- cooled liquids: Implications for establishing detailed con- nection between thermodynamic and kinetic anomalies in supercooled liquids. Journal of Chemical Physics, 119, Article ID: 9088. doi:10.1063/1.1614180 [25] Reichl, L.E. (1980) A modern course in statistical physics. University of Texas Press, Dallas. [26] Pathria, R.K. (1977) Statistical mechanics. Pergamon Press, Oxford. [27] Williams, G. and Watts, D.C. (1970) Non-symmetrical dielectric relaxation behavior arising from a simple em- pirical decay function. Transactions of the Faraday Soci- ety, 66, 80-85. doi:10.1039/tf9706600080 [28] Physics Survey Committal (1986) Physics through the 1990s, condensed-matter physics. National Academy, Washington. [29] Frisch, U. (1995) Turbulence. Cambridge University, Cambridge. [30] de Gennes, P.G. (1985) Scaling concepts in polymer physics. Cornell University, New York. [31] Lubchenko, V. and Wolynes, P.G. (2001) Intrinsic quan- tum excitations of low temperature glasses. Physical Re- view Letters, 87, 1-4. doi:10.1103/PhysRevLett.87.195901 [32] Kent, J.A. (2007) Riegel’s handbook of industrial chem-  J.-L. Wu / Natural Science 3 (2011) 580-593 Copyright © 2011 SciRes. OPEN ACCESS 593 istry and biotechnology. Vol. 1, 11th Edition, Springer, New York. [33] Frank, H.P. (1986) Polypropylene. Gordon and Breach Science, New York. [34] Zhao, D., Fan, Q., Quian, R. and Xiu, D. (1981) Rela- tionship between the zero shear rate viscosities of PP melt and its viscosity average molecular weights. Bulle- tin of Macromolecula, 5, 385-388. [35] Wright, W.B., Budakian, R., Pine, D.J. and Putterman, S.J. (1997) Imaging of intermittency in ripple-wave tur- bulence. Science, 278, 1609-1216. doi:10.1126/science.278.5343.1609 [36] Vassilicos, J.C. (2001) Intermittency in turbulence flows. Cambridge University, Cambridge, 44-63.
|