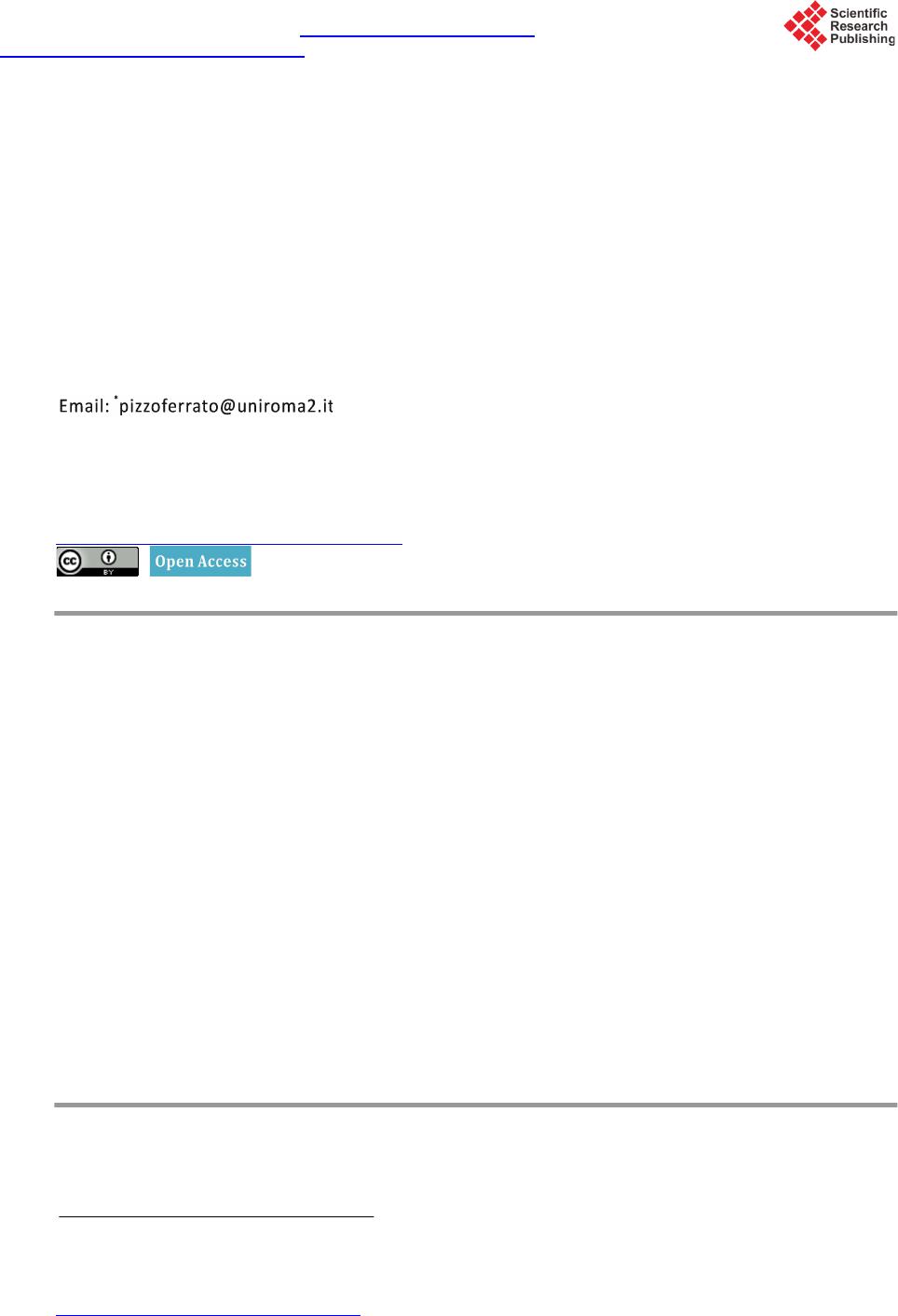
 Materials Sciences and Applications, 2015, 6, 943-952 Published Online November 2015 in SciRes. http://www.scirp.org/journal/msa http://dx.doi.org/10.4236/msa.2015.611095 How to cite this paper: Pizzoferrato, R., Tagliatesta, P., Schillaci, C., Prosposito, P. and De Angelis, R. (2015) Synthesis and Photophysical Properties of 9,10-Disubstituted Anthracenes. Materials Sciences and Applications, 6, 943-952. http://dx.doi.org/10.4236/msa.2015.611095 Synthesis and Photophysical Properties of 9,10-Disubstituted Anthracenes Roberto Pizzoferrato1*, Pietro Tagliatesta2, Carlo Schillaci2, Paolo Prosposito1, Roberta De Angelis1 1Dipartimento di Ingegneria Industriale, Università di Roma Tor Vergata, Roma, Italy 2Dipartimento di Scienze e Tecnologie Chimiche, Università di Roma Tor Vergata, Roma, Italy Received 11 September 2015; accepted 14 November 2015; published 17 Nove mber 2015 Copyright © 2015 by authors and Scientific Research Publishing Inc. This work is licensed under the Creative Commons Attribution International License (CC BY). http://creativecommons.org/licenses/by/4.0/ Abstract We report the synthesis and photophysical characterization of four 9,10-disubstituted dipheny- lanthracenes with specific modifications of the model backbone which involve both the 9,10 par a substituents at the phenyl rings and the substitution with carbon-carbon triple bonds. The effects of such modifications on the photoluminescence and electroluminescence properties have been investigated on the basis of the diphenylanthracene molecular characteristics and in view of ap- plication to light-emitting devices. We have found that the substitution with the carbon-carbon triple bonds at the two 9,10-phenyls noticeably alters the electronic states of the reference mole- cule, also introducing a certain degree of sensitivity to the phenyl substituents, which improves the tunability of the optical emission. Differently, the 9,10 para substituents produce minor changes in the single-molecule properties, due to the lack of electronic conjugation across the 9,10-phenyls. However, even a single nitro substituent in the phenyl para position produces the formation of ex- cimers, which appreciably reduces the optical quantum efficiency. These properties are main- tained in solid-state blends and simple spin-coated bilayer electroluminescent devices have been fabricated. Keywords Blue Mate rial , Electroluminescence, PVK, 9,10-Diphenylanthracene, Organic Li ght-Emitting Devices 1. Introduction In the last thirty years, anthracene derivatives have demonstrated to have a surprisingly great number of applica- Corresponding author.  R. Pizzoferrato et al. tions in very different fields, which range from anticancer agents [1] to fluorescence probes and chemical sen- sors [2] [3] and also encompassing thin-film transistors [4 ] and solar cells [5]. Since their first application in early organic light-e mittin g devices (OLE D) [6], the se co mp ound s have witnesse d a furthe r ri sing i nter e s t d ue to the great chance of molecular engineering with the addition of substituent groups [7]-[11] as well as with the exploitation of less standard process, such as the triplet-triplet annihilation [12] [13]. Synthesis and investiga- tions have especially been aimed to achieve efficient, pure and stable emission in the blue side of the spectrum as well as to a ccomplish t unability towards lo nger wavele ngths and good solubility for s pin-coating processing. Specifically, even though recent studies have addressed the 2,6-subst i tute d a nt hr ac enes [9], whic h a r e l es s p r one to face-to-face packing (π-π stacking) and consequent decrease of emission efficiency, the greatest interest has been risen by the 9,10-substituted anthracenes and especially by 9,10-disubstituted diphenylanthracene (DPA) derivatives [6] [7] [14]-[20], ba sin g o n t he 95 % q uant u m yie ld o f DP A itsel f a nd o n t he gr ea t c hanc e of mol ecu- lar engineering offered by its molecular structure. Within this framework, we ha ve s ynthes ized fo ur 9,1 0-disub- stituted diphenylanthracene derivatives with specific modifications of the model backbone, involving both 9,10 para substituents at the phenyl rings and the substitution with carbon-carbon triple bonds. The effects of such modifications on the photophysical properties and the chance of application to OLEDs have thus been investi- gated by photoluminescence (PL) measurements and by implementing simple bilayer electroluminescence (EL) devices. 2. Experimental Section 2.1. Material Synthesis The molecular str ucture of the four synthesized 9,10-disubstituted diphenyla nthracene derivati ves are shown in Figure 1. In this section, in add ition to t he pr epa ratio n of the d er iva tives, hereafter referred to as A, B, C and D, all the reaction i nter mediates are also reported. 9,10-bis-(4-methoxyphenyl)anthracene (A): 1 g (3.0 × 10−3 mol) of 9,10-dibromoanthracene was dissolved in 20 ml of toluene and 2.28 g (1.5 × 10−2 mol) of 4-methoxyphenyl boronic acid were added. To the obtained mixture, 20 mg of Pd(AcO)2, 40 mg of triphenylphosphine and 5 ml of a 1 M Na2CO3 solution were added. The mixture was deoxygenated with an argon stream for 20 min and refluxed for 18 h under nitrogen. The reaction was cooled at room temperature and 100 ml of water were added. The solution was twice extracted with diethyl ether and afte r the eva porat ion of the solve nt, the re sidue wa s puri fied by flash c hromatogr aphy (Si O2, 1% die- thyl e ther/he xane mixture) and the desired compound was identified by 1H NMR and mass spectra. Yield 60%. m.p. 272˚C - 273˚C; 1H NMR(400 MHz, CDCl3) δ = 7.73(m, 4H), 7.40(d, 4H, J = 12 H z), 7 .32(m, 4H), 7.14(d, 4H, J = 12 Hz), 3.96(s, 6 H): EI MS: m/z(%) 390(100). Figure 1 . Chemical structures of the DPA deri vat ives.  R. Pizzoferrato et al. 9-bromo-10-(4-methoxyp he nyl)anthrace ne : 1 g (3.0 × 10−3 mol) of 9,10-dibromoanthracene was dissolved in 20 ml of toluene and 1.14 g (7.5 × 10−3 mol) of 4-methoxyphenyl boronic acid were added. To the obtained mixture, 20 mg of Pd(AcO)2, 40 mg of triphenylphosphine and 5 ml of a 1 M Na2CO3 solution were added. The mixture was deoxygenated with an argon stream for 20 min and refluxed for 18 h under nitrogen. The reaction was cooled at room temperature and 100 ml of water were added. The solution was twice extracted with diethyl ether and afte r the eva porat ion of the solve nt, the re sidue wa s puri fied by flash c hromatogr aphy (Si O2, 1% die- thyl e ther/he xane mixture) and the desired compound was identified by 1H NMR and mass spectra. Yield 80%. m.p. 187˚C - 188˚C; 1H NMR (400 M Hz, CDCl3) δ = 8.62(d, 2H, J = 8.2Hz), 7.67(d, 2H, J = 8.2Hz), 7.59(t, 2H, J = 6.5Hz), 7.38(t, 2H, J = 6.5Hz), 7.53(d, 2H, J = 8.1Hz), 7.14(d, 2H, J = 8.1Hz), 3.98(s, 3H); EI MS: m/z(%) 363(100). 9-(3-nitrophenyl)-10-(4-methoxyphenyl)anthracene (B): 1 g (2.8 × 10−3 mol) of 9-bromo-10-(4’-metho- xyphenyl)anthracene was dissolved in 20 ml of toluene and 1.4 g (8.4 × 10−3 mol) of 3-nitrophenyl boronic acid was added. To the obtained mixture, 20 mg of Pd(AcO)2, 40 mg of triphenylphosphine and 5 ml of a 1 M Na2CO3 solution were added. The mixture was deoxygenated with an argon stream for 20 min and refluxed for 18 h und er nitro ge n. T he reaction was cooled at room temperature and 100 ml of water was added. The solution was twice extracted with diethyl ether and after the evaporation of the solvent, the residue was purified by flash chro matogr aphy ( SiO2, 5% die thyl et her/he xane mixtur e) and the desired compound was identified by 1H NMR and mass spectra. Yield 60%. m.p. 301˚C - 302˚C; 1H NMR(400 MHz, CDCl3) δ = 8.49(d, 2H, J = 12.2 Hz), 7.77(m, 2H), 7.32(m, 2H), 7.67(d, 2H, J = 12.2 Hz), 7.53(m, 6H), 7.17(d, 2 H, J = 12.2 Hz), 3.97(s, 3H): EI MS: m/z(%) 405(100). 9,10-bis-(3-nitrophenyl)anthra c e ne : 1 g (3.0 × 10−3 mol) of 9,10-dibromoanthracene was dissolved in 20 ml of toluene and 2.5 g (1.5 × 10−2 mol) of 3-nitrophenyl boronic acid were added. To the obtained mixture, 20 mg of Pd(AcO)2, 40 mg of triphenylphosphine and 5 ml of a 1 M Na2CO3 solution were added. The mixture was deoxygenated with an argon stream for 20 min and refluxed for 36 h under nitrogen. The reaction was cooled at room temperature and 100 ml of water were added. T he solution was twice extracted wit h chloroform and after the evaporation of the solvent, the residue was purified by flash chromatography (SiO2, 10% diethyl ether/hex- ane mixture) and the desired compound was identified by 1H NMR and mass spectra. Yield 40%. m.p. >320˚C; 1H NMR(4 00 MHz, CDCl3) δ = 8.51(d, 4H, J = 12.4 Hz), 7.69(d, 4H, J = 12.4 Hz), 7.58(m, 4H), 7.41(m, 4H); EI MS: m/z(%) 420(100). 9,10-bis-[(4-methoxyphenyl)ethynyl]anthracene (C): 1 g (3.0 × 10−3 mol) of 9,10-dibromoanthracene was dissolved in 20 ml of dry THF and 6.5 g (1.5 × 10−2 mol ) o f 4 -ethynyl-methoxybenzene were added. To the ob- tained mixture, 0.2 ml of 10% tetrabutylammonium fluoride THF solution was added. The homogeneous solu- tion was deoxygenated with an argon stream for 20 min after that 20 mg of Pd(PPh3)2Cl2 were added under ni- trogen. The mixture was further deoxygenate with an argon stream for 20 min and kept at 60o C for 12 hours. The reaction was cooled at room temperature and 100 ml of water were added. T he solution was twice extracted with chloroform and after the evaporation of the solvent, the residue was purified by flash chromatography (SiO2, 30% dichlomethane/hexane mixture) and the desired compound was identified by 1H NMR and mass spectra. Yield 50%. m.p. 230˚C - 232˚C; 1H NMR (400 MHz, CDCl3) δ = 8.70(dd, 4H, J = 3.4 Hz), 7.73(d, 4H, J = 9.2 Hz), 7.65(dd , 4H, J = 3.4 Hz), 7.0(d, 4H, J = 9 Hz); EI MS: m/z(%) 438(100). 9,10-bis-[(2,6-dimethoxyphenyl)ethynyl]anthracene (D): 1 g (3.0 × 10−3 mol) of 9,10-dibromoanthracene was dissolved in 20 ml of dry THF and 7.47 g (1.5 × 10−2 mol) of 2,6-dimethoxy-ethynylbenzene [21] were added. To the obtained mixture, 0.2 ml of 10% tetrabutylammonium fluoride THF solution was added. The ho- mogeneous solution was deoxygenated with an argon stream for 20 min after that 20 mg of Pd(PPh3)2Cl2 were added under nitrogen. The mixture was further deoxyge nate with an argon stream for 20 min and kept at 60o C for 12 hours. The reaction was cooled at room temperature and 100 ml of water were added. The solution was twice extracted with chlorofor m and after the evapora tion of the solvent, the residue was purified by flash chro- matography (SiO2, 60% dichlomethane/hexane mixture) and the desired compound was identified by 1H NMR and mass spectra. Yield 60%. m.p. 301˚C - 302˚C; 1H NMR(400 MHz, CDCl3) δ = 8.89(m, 4H), 7.63(m, 4H), 7.29(t, 2H, J = 2.4 Hz)), 6.61(d, 4H, J = 1.6 Hz), 4.07(s, 12 H),: EI MS: m/z(%) 498(100). 2.2. Photophysical Measurements For the optical measurements in liquid solutions, spectroscopic-grade tetrahydrofuran (THF) was used as re- 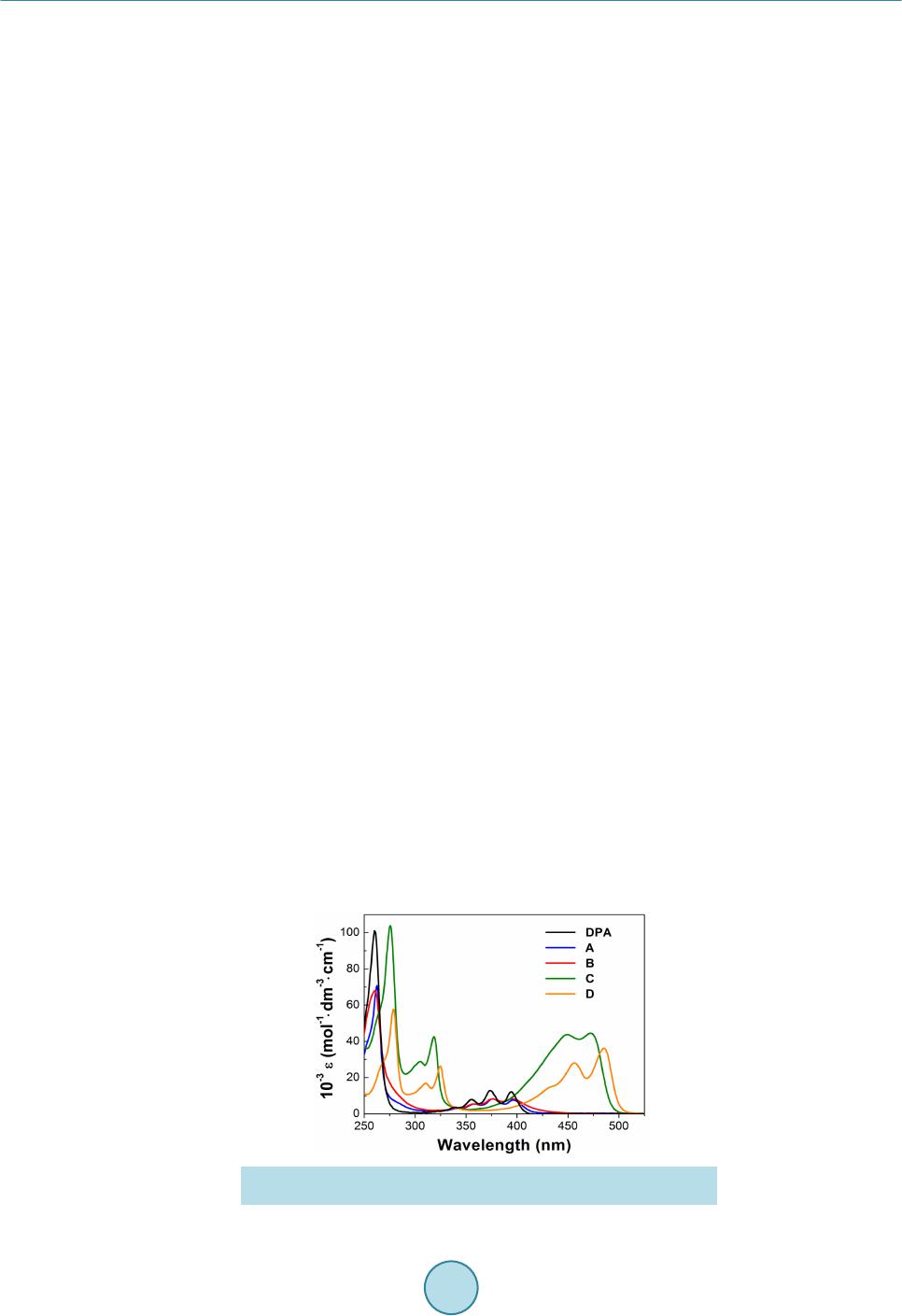 R. Pizzoferrato et al. ceived from Sigma-Aldrich. UV-vis spectra were recorded with a Cary 50 (Varian Inc.) spectrophotometer: di- luted solutions were held in silica cells with path lengths of 10 mm and the spectra recorded over the range 200 - 600 nm with a 0.5 nm data interval and a 1 nm band-pass. Fluorescence spectra were recorded on a standard la- boratory set-up for PL measurements equipped with a photo-multiplier (Ha mamatsu R38 96 ) p laced at the output slit of a 25-cm monochromator (Oriel Cornerstone 260). Provided the diluted solutions (typical molar concen- tration of 10 −6 M) had an absorbance of <0.05 in the excitation range, the fluorescence spectra were recorded by exciting the sample with the conventional 90˚ geometry and the monochromatized output of a Hg-Xe cw dis- charge lamp. Both the excitation and emission spectra were fully corrected by using the se t-up cal ibr atio n cur ve obtained with a reference black-body lamp and standard fluorophore diluted solutions. A spectral band pass of 2 nm was used for both the excitation and emission monoc hro mators. For the EL measurements, simple bilayer devices were prepared as follows. Devices were spin-coated on glass/ITO substrates (Ossila). ITO had a typical thickness of 100 nm and a surface resistivity of 20 Ω/sq. The substrates were carefully cleaned with a 10% solution of NaOH in water at 90˚C in an ultrasonic bath. A Poly [3,4-ethylenedioxythiophene]:Poly[styrenesulphonic acid] (PEDOT:PSS) layer was previously spin-coated on the ITO surface as hole transport layer (HTL). The emissive layer (EML) was composed by the DPA substituted species dissolved in a blend of Poly(9-vinylcarbazole) (PVK) and 2-[4-tert-Butylphenyl]-5-[4-biphenylyl]-1,3,4- oxadiazole) (Butyl-PBD) purchased by Sigma Aldr ich and use d witho ut furt her purification. Bu tyl-PB D acts as electron transport layer (ETL) and plays a fundamental role in enhancing EL. PVK and Butyl-PBD were dis- solved in spectroscopic grade chloroform at a concentration of 20 mg/ml and 10 mg/ml, respectively. DPA d e- rivatives were added with a relative concentration of 5% by weight with respect to the PVK content. Doped PVK/Butyl-PBD layer was spin-coated on PEDOT:PSS layer at 3000 rpm for 45 s and densified in vacuum at 120˚C for 10 min. As a final step a thin layer of about 100 nm of aluminum was evaporated under vacuum (<1 × 10−6 Torr) on the outer surface of the film as cathode. All fabrication processes were carried out in clean room and in controlled atmosphere. EL measurements were performed in air with a relative humidit y in the range 60% - 65% without any sealing protection. PL measurements in solid-state films were carried out on the same sam- ples as for EL, but without the deposition of the aluminum cathode. 3. Results and Discussion 3.1. Absorption Spectra Figure 2 compares the molar absorptivity spectra of the different compounds in diluted THF solutions (molar conc ent ra tio n 10 −6 M). It is interesti ng to discus s the spectra of the differe nt derivatives b y considerin g the respec- tive modifications made to the reference DPA molecule along the axis linking the two 9,10-phen yls. It is, in fac t, well established [3] [7] [8] [9] [18] [22] that the lower-ener gy UV pr ogre ssio ns of b ot h ant hra cene a nd DPA a re determined by an allowed optical transition S0 → S1, assigned to A1g → B2u in group t heor et ical notat io n, wit h a dipole moment lying along the short axis of the molecule backbone. However, in the present compounds, neither type of substitution at the para position of 9,10-phenyls, i .e. the methoxy group in compound A or the nitro group in B, see ms to prod uce sig nificant varia tions and the spectra o f the two derivatives c lo sel y trace that of the reference unsubstituted DPA (Figure 2). This occurs with respect to both the typical lower-energy progression Figure 2. Absorptivity spectra of the different compounds in diluted THF solutions (molar concentration 10−6 M).  R. Pizzoferrato et al. in the r an ge 3 5 0 - 400 n m a nd t he hi ghe r -energy peak at 260 nm due to S0 → S2 transitions ( A1g → B3u). A s i mi- lar insensitivi ty to 9, 10 para substituents wa s already reported in non-s ym met ri c D PA de ri vat i ves wi th r e ga rd t o the allowed S0 → S1 transition [7] [10] and is in agreement with theoretical calculations showing that only a small fractio n of molecular o rbitals extends to wards the sub stituents [7] [19]. W e sugge st that t he to rsion o f the single carbon-carbon bond due to the right-angle twisting of the two 9,10-phenyls [7] is also effective in re- straining the conjugation within the anthracene backbone and preventing it from involving the different para substituents. Conversely, quite noticeable effects are produced by the substitution of the carbon-carbon triple bonds for the single bonds linking the two 9,10-phenyls, which is performed in C and D. The higher-energy peak exhibits a bathochromic shift from 260 nm to approximately 275 nm and, more significantly, the lower-energy progression is apparently split in two groups, centred at 310 and 450 nm, respectively, which exhibit a fourfold increase of the mol ar ab so r p tivi t y i n c o mp ar is on wit h DPA, wit h va l ue s a s hi g h as ε ≅ 4 × 104 M−1∙cm−1. A similar splitting, with the arising of two well defined progressions centred at 325 and 390 nm, was observed in 2,6 substituted anthracene derivatives [9] and attributed to a mixing of the two lo w-lyi ng excite d sta te s La and Lb of polyacenes, with the reorientation of the dipole moments and remodulation of the respective oscillator strength. Moreover, the spectra of C and D display a certain dependence on the type and positions of the 9,10-phenyls substit uents, with D exhibiting an appreciably higher bathochromic shift. All these findings show the greater effectiveness of the carbon-carbon triple bond in modifying the charge distribution of the anthracene backbone, also by allowing a greater extension of the molecular orbitals towards the two 9,10-phenyls due to the symmetry of triple bond against rotatio nal motion s. This could i mprove the π-conjuga tion t hus e xpla ini ng t he ge ner al b atho chro mic s hift of the spectral structures in C and D, as well as the sensitivity to the 9,10 -phenyls substitue nt s . In all the compounds A-D, the molar absorptivity remained constant and the absorption curves did not show any change up to values of the molar concentration as high as 10−3 M, wh ich r ules o ut the possible formation of aggregates. 3.2. Photoluminescence Spectra Figure 3 displays the photoluminescence (PL) spectra of the different derivatives in d ilut ed THF solution s (10 −6 M) excited at λ ex = 340 nm while the values of the relevant photophysical parameters are reported in Table 1. The differences in the emission spectra largely reflect those of the absorption curves, sho win g t ha t t he molec ular relaxation doe s not dra maticall y alter the different charac ters of the spatial distrib ution of e lectron densit y in the excited states. In fact, the spectra A and B are similar to that o f the unsubstituted D PA, apart fro m a slightl y la r- ger St oke s ’ shift and the smearing of the vib ronic bands with the comple te lack of the mir ror i mage effect. Thes e minor differences indicate a more effective intramolecular conformational relation, which leads to slightly dif- ferent relaxed excited states (RES) and cou ld origin fro m a slig htly greater flexibili ty of the molecule due to the para substitue nts o f the 9, 10-phenyls. On the other hand, the spectra of C and D are quite different from that of the unsubstituted DPA and are very similar to each other, with a large a bathochromic shift resulting in a main peak at 485 and 490 nm, respectively, and a smaller one around 520 nm. Similar to the case of absorption, these characteristics resemble the results reported in 2,6 substituted anthracene derivatives [9], thus confirming that the carbon-carbon triple bonds effectively alter the electronic states of the anthracene core, also by improving the conjugation along the 9,10-phenyl axis. Table 1. Absorption pea k wav e leng t h, mola r a bs or pti v it y, fluore s ce nc e pe a k wav e le ngth (in s ol uti o n and solid sta t e film ) a nd quantum yields of DPA derivative A-D. Comp ound Absmax (nm)a logε (l mol−1∙cm−1)a PLmax solution a (n m ) PLmax sol id state b (nm) Q.Y. A 263 4.84 420 426 0.45 B 261 4.83 431 435 0.20 C 275 5.01 485 492 0.70 D 278 4.76 492 502 0.60 aRecorded in 10−6 mol∙L−1 THF solution; brecorded in 5% blends with PVK.  R. Pizzoferrato et al. Figure 3. PL spectra of the different derivatives in diluted THF solutions (10−6 M) ex cited at λ ex = 340 nm. Moreover, when C and D are excited at either the absorption group centred at 310 and the one at 450 nm, re- spectively, the same PL spectra around 500 nm are observed indicating that the carbon-carbon triple bonds still allow an effective intramolecular relaxation. With regard to A and B, however, even though the para substitu- ents d o no t s i gn ific a ntl y a l te r t he RES, thus the spectra of diluted solutions, it should be noted that the nitro sub- stituent in B produces some relevant effects on the excited state, as we have found by increasing the molarity of solution. In fact, while the PL signal of DPA, A, C, a nd D showed a linear increase of intensity with concentra- tion and a constant spectral profile, the spectrum of B featured the typical behaviour of the formation of exci- mers, similar to pyrene in cyclohexane [23]. Specifically, as reported in Figure 4, at molarity higher than 5 × 10−6 M a broad emission band appears with the peak at 600 nm and grows with increasing concentration at the expe nse of t he si n gle -molecule e mission (430 nm) which decreases with concentration. Differently from pyrene, in B the formation of excimers, through the π-staking dimerization of two molecules, seems to originate from the peculiar electron-withdrawing nature of the nitro group, which produces two complementary effects [24]. First it withdraws significant electron density from the acene π-cloud, thus decreasing the repulsion of the π-faces. Second, it creates a significant electric dipole and favours the molecular alignment due to dipole-dipole interaction. Finally, it must pla y a determinant ro le also in the fact tha t B exhibits the lowest value of low-con- centration quantum efficiency among all the derivatives (see Table 1), while A is mainly affected by the more effective relation processes in comparison to DPA, thus exhibiting half the quantum yield of the reference molecule. From this point of view, as well, the substitution with the carbon-carbon triple bond produces a no- ticeable effect by appreciably increasing the quantum yield of C and D, beyond the va lue of A. 3.3. Solid Films and Electroluminescence As preliminary step towards the implementation of an electroluminescent device, we investigated the photo- physi cal propert ies of thin films of PVK blended with the DPA derivatives. Figure 5 illustrates the PL spectra of a PVK fi lm doped w ith diff erent w eight conc entrat ions of A u nder ex cita tion at 330 nm . The curves are normalized by the absorption of the respective sample, so that they can be compared to each other and the PL i ntensity is an estimate of the relative quantum efficiency. The behaviour of the spectra with the concentration of A is typical of an energy transfer process, presumably based on a Forster resonan t mechanism, since the emi ssion band of PVK at 400 nm i s rapidly quench ed by small quantit ies of gues t, while t he PL of A a t 430 nm prog ressively incre ases with the concentration. In fact, while the excitation light is absorbed by both the host and guest species, the relative emission of the guest A should be negligible, due to the much lower concentration in comparison to PVK, unless a noticeable energy transfer occurs from PVK to A. We note that t his effect co mbines the la rge absorption of the PVK with the higher efficiency of the guest, thus effectively increasing the quantum efficiency of the film by a factor of four, approximately. Beyond a concentration of 1% by weight, the emission of A begins to saturate and eventually decreases due t o the arising of intermolecular concentration effects. Finally, we note that the lineshape and peak po sition (428 nm) of PL in the solid state ar e very similar to those in diluted solutions (see Figure 3), which confirms the small imp ortance of torsional motions in the relaxation processes of DPA derivatives [7]. By using the optimal weight ratio of 5%, we fabricated simple bilayer electroluminescent devices with deriva- tives A, C and D, as described in paragraph 2.2. B was not used since, as expected, it exhibited a very low 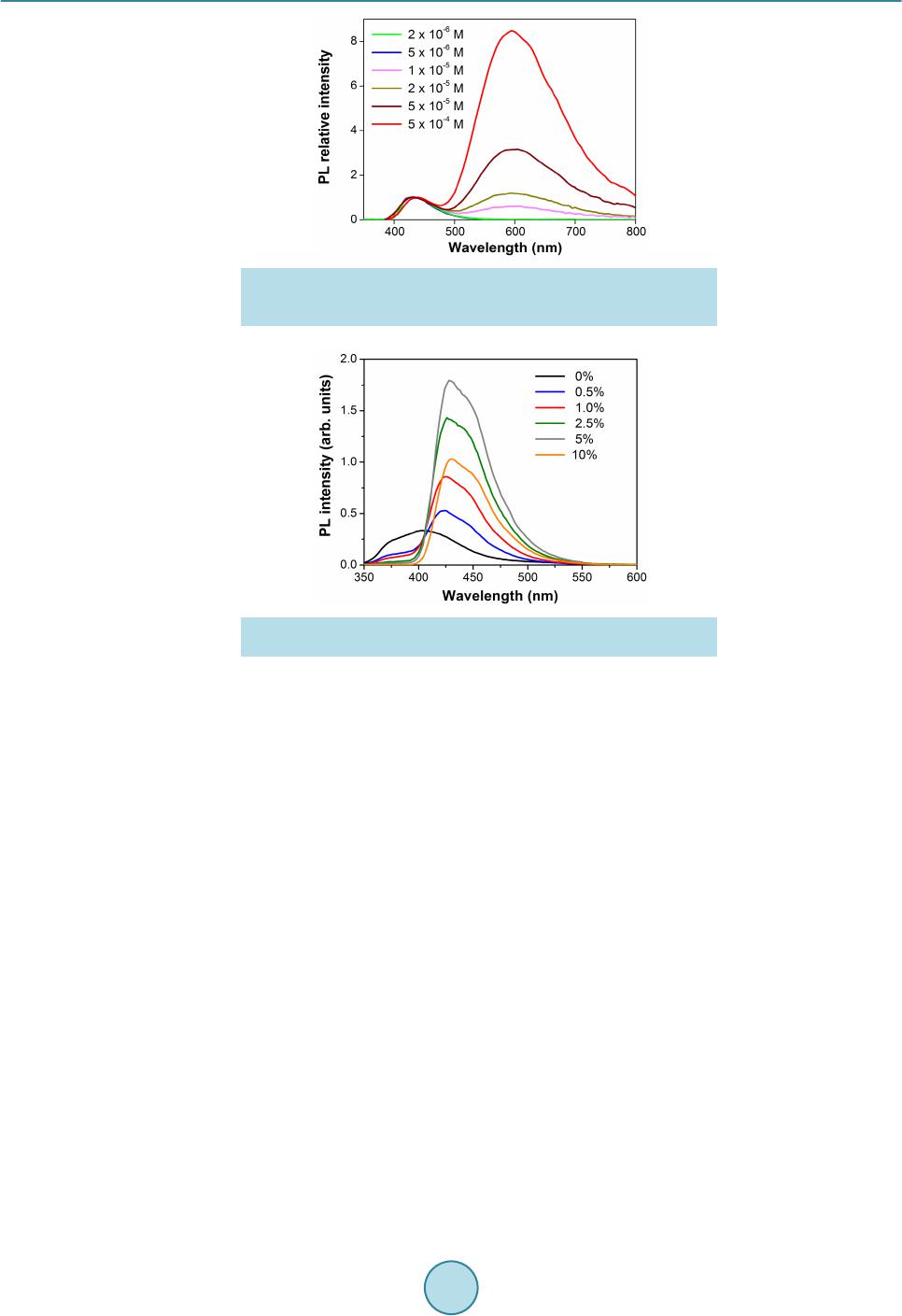 R. Pizzoferrato et al. Figure 4. PL intensity of B in THF solutions as a function of the molar concentration. The curves are normalized at the maximum of the single-molecule emission (430 nm). Figure 5. PL spectra of a PVK thin film doped with different con- centrations of the derivative A under excitation with light at 330 nm. efficiency in the solid state due to its tendency to form excimers. Figure 6 reports the EL spectrum of a device made with a PVK:A = 95:5 doped film along with the EL signal of a neat-PVK de vice. T he EL emis sio n of t he doped film has a maximum at 425 nm and is quite similar to the PL of A both in so lutio n and a s a gues t in P VK film (see Figure 5), with the lack of the low-energy tail of the PVK emission. This indicates that an efficient energy transfer from PVK to A is still effective under an electrical excitation, even though the turn-on voltage was foun d to be rel ativ ely h igh at 15 V while the variation of applied voltage had no ef fect on the sh ape and the peak of the EL spectra. The slightly different profile of EL, compared to PL, underlines the different nature of the t wo processes and the consequent related effects. For example, it should be considered the possible presence of spe- cific quenching sites for excitons in the EL device, due to the presence of an evaporated metal cathode. Figure 7 reports the EL spectrum of a device made with a 5% PVK:D doped film. The PL emission from a similar thin film deposited on ITO-coated glass, witho ut the alumin ium e lectro de, is also sho wn for co mpariso n. We note the presence of a very effective energy transfer mechanism, both in PL and EL, since there is no trace of the emission from PVK, si milar to the case of PVK:A. M oreover, the PL spectru m in the solid state is virtu- ally identical to that of dil uted liq uid solution (Fig ure 3). This indicate s that the molecula r relaxatio n processes, which are responsible for the large Stoke shift, do not involve the tor sional motions of the 9, 10-phenyl moieties, as it also occurs in unsubstituted DPA [7], even in the presence of the carbon-carbon triple bonds. In this case, however, t he sp e ctral profile o f EL is very si milar to that of PL but with a uni form shift to longer wavele ngt hs of 10 nm, approximately. This effect, already observed in organic emitters [25] [26], is again due to the different nature of the two processes and could mainly origin from at least two mechanisms. First, differently from PL, in EL the injected charges migrate through the sample and will naturally move to lower energy sites even before exciton formation. Second, EL also generates triplet excitons thus directly populating the triplet states of the emitter, with t he emission o f red -shifted phosphorescence. Similar results were also reported for 5% PVK:C de- vices (not shown).  R. Pizzoferrato et al. Figure 6. EL spectrum of a device made with a 5% PVK:A do ped film along with the EL of a PVK neat-film device. Figure 7. EL and PL spectra of a device made with a 5% PVK:D doped film. 4. Conclusion We have synthesized four 9,10-disubstituted diphenylanthracenes that emit in the blue-green region. The mod- ifications made to the reference DPA backbone reflect on the photophysical properties of the derivatives in a way that can easily be understood on the basis of the electronic characteristics and demonstrate the versatility of the DPA backbone. Specifically, the addition of para substituents to the 9,10 phenyl rings does not significantly alter the absorption and the emission spectra of DPA, in agreement with the hypothesis that the electronic con- jugation does not extend across the 9,10 phenyl rings, even though the quantum yield is appreciably reduced due to the increase of the molecular flexibility and the number of relaxation mechanisms. However, the electron- withd ra wing nat ur e o f the ni tro gr o up pr o d uce s the fo r mat io n o f e xc i mer s whe n t he mol ar co nce ntr at ion in so lu- tion is increased beyond 10−5 M, whic h clearl y preven ts the us e in so lid-state light e missio n devices. Quite dif- ferently, the substitution of the carbon-carbon triple bonds for the single bonds linking the two 9,10-phenyls produces relevant effects on the electronic states, apparently by extending the conjugation across the phenyl rings. In add ition to a noteworthy red shift of e mission towards the green regio n, this increases the quantum ef- ficiency and introduces a certain degree of sensitivity to the 9 ,10 phen yl subs tituent s, which i mproves t he tuna- bility of the optical emission. The overall stiffness of the DPA backbone, even in the presence of the car- bon-carbon triple bonds, ensures that these properties are substantially maintained in the solid state. Therefore, by exploiting an efficient energy transfer mechanism in blends with PVK as a host, the derivatives have been used to fabricate simple bilayer electroluminescent devices which exhibit a blue emission with a greater color purity and red-shifted bands in the blue-green region. We believe these results demonstrate that diphenylan- thracenes, d espite their simple structure and quite lon g-stand ing s tor y of in vestigatio ns, are still far fro m being a depleted field for molecular engineering and development of new emitting materials. In particular, the findings about the role of the carbon-carbon triple bonds provide useful suggestions for future research and applications to OLEDs.  R. Pizzoferrato et al. Acknowledgements We thank Riccardo De Gennaro for his support and assistance during the preparation and characterization of electroluminescent devices. References [1] Agbandje, M., Jenkins, T.C., McKenna, R., Reszka, A.P. and Neidle, S. (1992) Anthracene-9,l O-Diones as Potential Anticancer Agents. Synthesis, DNA-Binding, and Biological Studies on a Series of 2,6-Disubstituted Derivatives. Journal of Medicinal Chemistry, 35, 1418-1429. http://dx.doi.org/10.1021/jm00086a010 [2] Becker, H.D. (1993) Unimolecular Photochemistry of Ant hracenes. C hemical Reviews, 93, 145-172. http://dx.doi.org/10.1021/cr00017a008 [3] V aleur, B. (2002) Molecular Fluorescence: Principles and Applications. Wiley-V C H , Weinheim. [4] Meng, H., Sun, F.P., Goldfinger, M.B., Jaycox, G.D., Li, Z.G., Marshall, W.J. and Blackman, G.S. (2005) High-Per- formance Stable Organic Thin-Film Field-Effect Transistors Based on Bis-5’-alkylthiophen-2’-yl-2,6-Anthracene Se mi- conductors. Journal of the American Chemical Society, 127, 2406-2407. http://dx.doi.org/10.1021/ja043189d [5] Teng, C., Yang, X., Yang, C., Li, S., Cheng, M., Hagfeldt, A. and Sun, L. (2010) Molecular Design of Anthracene- Bridged Metal-Free Organic Dyes for Efficient Dye-Sensitized Solar Cells. Journal of Physical Chemistry C, 114, 9101-9110. http://dx.doi.org/10.1021/jp101238k [6] Shi, J. and Tang, C.W. (2002) Anthracene Derivatives for Stable B lue-Emittin g Organ ic El ectro lu min escen ce De vices. Applied Physics Letters, 80, 3201-3203. http://dx.doi.org/10.1063/1.1475361 [7] Serevičius, T., Komskis, R., Adomėnas, P., Adomėnienė, O., Jankauskas, V., Gruodis, A., Kazlauskas, K. and Juršėnas, S. (2014) Non-Symmetric 9,10-Diphenylanthracene-Based Deep-Blue Emitters with Enhanced Charge Transport Pro- perties . Physical Chemistry C hemical Physics, 16, 7089-7101. http://dx.doi.org/10.1039/c4cp00236a [8] Lin, S.H., Wu, F.I. and Liu, R.S. (2009) Synthesis, Photophysical Properties and Color Tuning of Highly Fluorescent 9,10-Disubstituted-2,3,6,7-Tetraphenylanthracene. C hemical Communications, 6961-6963. http://dx.doi.org/10.1039/b912289c [9] Zhou, X., Piland, G.B., Kurunthu, D., Dillon, R.J., Burdett, J.J. and Bardeen, C.J. (2012) The Photophysics of the 2,6 Dialkoxy Anthracenes: Evidence for Excited State Side-Chain Conformational Relaxation. Journal of Luminescence, 132, 2997-3003. http://dx.doi.org/10.1016/j.jlumin.2012.06.012 [10] Mallesh am, G., Balaiah, S., Ananth Reddy, M., Sridhar, B., Singh, P., Srivastava, R ., Bhanuprakash, K . and Jayathirtha Rao, V. (2014) Design and Synthesis of No vel Anthracene Deri vatives as n-Type Emitters for E lectrolu minescent De- vices: A Combined Experimental and DFT Study. Photochemical & Photobiological Sciences, 13, 3 4 2-357. http://dx.doi.org/10.1039/c3pp50284h [11] Kim, R., Lee, S., Kim, K.H., Lee, Y.J., Kwon, S.K., Kim, J.J. and Kim, Y.H. (2013) Extremely Deep Blue and Highly Efficient Non-Doped Organic Light Emitting Diodes Usin g an Asymmetric Anth racen e Deri vativ e with a X ylene Un it . Chemical Communications, 49, 4664. http://dx.doi.org/10.1039/c3cc41441h [12] Fukagawa, H., Shimizu, T., Ohbe, N., Tokito, S., Tokumaru, K. and Fuj ikake, H. (2 012 ) Anthr acene Der ivatives as Ef- ficient Emitting Hosts for Blue Organic Light-Emitting Diodes Utilizing Triplet-Triplet Annihilation. Organic Elec- tronics, 13, 1197-1203. http://dx.doi.org/10.1016/j.orgel.2012.03.019 [13] Chiang, C.J., Kimyonok, A., Etherington, M.K., Griffiths, G.C., Jankus, V., Turksoy, F. and Monkman, A.P. (2013) Ultrahigh Efficiency Fluorescent Single and Bi-Layer Organic Light Emitting Diodes: The Key Role of Triplet Fusion. Advanced Functional Materials, 23, 739-746. http://dx.doi.org/10.1002/adfm.201201750 [14] Wu, C.L., Chang, C.H., Chang, Y.T., Chen, C.T., Chen, C.T. and Su, C.J. (2014) High Efficiency Non-Dopant Blue Organ ic Li ght-Emitting Diodes Based on Anthracene-Based Fluorophores with Molecular Design of Charge Transport and Red-Shifted Emiss ion Pr oof. Journal of Materials Chemistry C, 2, 7188 -7200. http://dx.doi.org/10.1039/C4TC00876F [15] Jo, W.J., Kim, K.H., No, H.C., Shin, D.-Y., Oh, S.J., Son, J.H., Kim, Y.H., Cho, Y.K., Zhao, Q.H., Lee, K.H., Oh, H.Y. and Kwon, S.K. (2009) High Efficient Organic Light Emitting Diodes Using New 9,10-Diphenylanthracene Deriva- tives Containing Bulky Substituents on 2,6-Positon. Synthetic Met als, 159, 1359-1364. http://dx.doi.org/10.1016/j.synthmet.2009.03.007 [16] Kim, S.K., Yang, B., Ma, Y., Lee, J.H. and Park, J.W. (2008) Exceedingly Efficient Deep-Blue Electroluminescence from New Ant hracenes Obtained Using Rational Molecular Design. Journal of Materials Chemistry C, 18, 3376-3384. http://dx.doi.org/10.1039/b805062g [17] Bin, J.K. and Hong, J.I. (2011) Efficient Blue Organic Light-Emitting Diode Using Anthracene-Derived Emitters Based on Polycyclic Aromatic Hydrocarbons. Organi c Electroni cs, 12, 802-808.  R. Pizzoferrato et al. http://dx.doi.org/10.1016/j.orgel.2011.02.011 [18] Ting, C.H. (1967) Electronic Structure and Intersystem Crossing in 9,10-Diphenylanthracene. Chemical Physics Let- ters, 1, 335-336. http://dx.doi.org/10.1016/0009-2614(67)80010-1 [19] Park, J.W., Kim, Y.H., Jung, S.Y., Byeon, K.N. and Jang, S.H. (2008) Efficient and Stable Blue Organic Light-E mi tting Diod e B as ed on an Anthr acene Derivative. T hin Solid Films, 516, 8381-8385. http://dx.doi.org/10.1016/j.tsf.2008.04.080 [20] Chien, C.H., Chen, C.K., Hsu, F.M., Shu, C.F., Chou, P.T. and Lai, C.H. (2009) Multifunctional Deep-Blue Emitter Comprising an Anthracene Core and Terminal Triphenylphosphine Oxide Groups. Advanced Functional Materials, 19, 560-566. http://dx.doi.org/10.1002/adfm.200801240 [21] All en, A. D . and Cook, C.D. (1963) Substituted Phenylacetylenes. Infrared Spectra. Canadian Journal of Chemistry, 41, 1084-1087. http://dx.doi.org/10.1139/v63-155 [22] Kukhta, A.V., Kukhta, I.N., Kukhta, N.A., Neyra, O.L. and Meza, E. (2008) DFT Study of the Electronic Structure of Anthracene Derivatives in Their Neutral, Anion and Cation Forms. Journal of Physics B: Atomic, Molecular and Opt- ical P hysics, 41, Article ID: 205701. http://dx.doi.org/10.1088/0953-4075/41/20/205701 [23] Birks, J.B. (1975) Excimers. Reports on Progress in Physics, 38, 903-974. http://dx.doi.org/10.1088/0034-4885/38/8/001 [24] Anthony, J.E. (2007) Induced π -Stacking in Acenes. In: Müller, T.J.J. and Bunz, U.H.F., Eds., Functional Organic Materials: Syntheses, Strategies and Applications, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, 511-542. http://dx.doi.org/10.1002/chin.200735267 [25] Lee, K.-S. (2002) Polymers for Photonics Applications I. Spring-Verl ag B er lin Heidelberg, New Yor k. [26] Ullaa, H., Garudacharib , B., Satyan arayana, M.N. , Umesha, G. and Isloorb, A.M. (2014) Blue Organic Light Emitting Materials: Synthesis and Characterization of Novel 1,8-Naphthalimide Derivatives. Optical Materials, 36, 704-711. http://dx.doi.org/10.1016/j.optmat.2013.11.017
|