 Advances in Microbiology, 2015, 5, 807-816 Published Online November 2015 in SciRes. http://www.scirp.org/journal/aim http://dx.doi.org/10.4236/aim.2015.512085 How to cite this paper: Watanabe, K., Horinishi, N. and Matumoto, K. (2015) Antibiotic-Resistant Bacterial Group in Field Soil Evaluated by a Newly Developed Method Based on Restriction Fragment Length Polymorphism Analysis. Advances in Microbiology, 5, 807-816. http://dx.doi.org/10.4236/aim.2015.512085 Antibiotic-Resistant Bacterial Group in Field Soil Evaluated by a Newly Developed Method Based on Restriction Fragment Length Polymorphism Analysis Katsuji Watanabe*, Naoto Horinishi, Kunimasa Matumoto Department of Life, Environment and Materials Science, Fukuoka Institute of Technology, Fukuoka, Japan Received 19 October 2015; accepted 13 November 2015; published 16 November 2015 Copyright © 2015 by authors and Scientific Research Publishing Inc. This work is licensed under the Creative Commons Attribution International License (CC BY). http://creativecommons.org/licenses/by/4.0/ Abstract Spreading of antibiotic resistant bacteria into environment is becoming a major public health problem, implicating affair of the indirect transmission of antibiotic resistant bacteria to human through drinking water, or vegetables, or daily products. Until now, the risk of nosocomial infec- tion of antibiotic resistant bacteria has mainly been evaluated using clinical isolates by phenotypic method. To evaluate a risk of community-acquired infection of antibiotic resistant bacteria, a new method has been developed based on PCR-RFLP without isolation. By comparing restriction frag- ment lengths of the 16S rDNA gene from bacterial mixture grown under antibiotic treatment to those simulated from the DNA sequence, bacterial taxonomies were elucidated using the method of Okuda and Watanabe [1] [2]. In this study, taxonomies of polymyxin B resistant bacteria group in field soils, paddy field with organic manure and upland field without organic manure were es- timated without isolation. In the both field soils, the major bacteria grown under the antibiotic were B. cereus group, which had natural resistance to this antibiotic. In field applied with organic manure, Prevotella spp., and the other Cytophagales, which were suggested to be of feces origin and to acquire resistance to the antibiotic, were detected. When numbers of each bacterial group were roughly estimated by the most probable number method, B. cereus group was enumerated to be 3.30 × 106 MPN/g dry soil in paddy field soil and 1.32 × 106 MPN/g dry soil in upland filed. Pre- votella spp. and the other Cytophagales in paddy field were enumerated to be 1.31 × 106 MPN, and 1.07 × 106 MPN∙g−1 dry soil. Keywords Polymyxin B Resistant Bacteria, Field Soil, Microchip Electrophoresis, Multiple Enzyme *Corresponding a uthor.  K. Watanabe et al. Restriction Fragment Length Polymorphism Analysis, the Most Probable Number Method 1. Introduction At present, various kinds of manures and compost originated from diverse biological wastes e.g., livestock feces, food waste, and sewage sludge, were introduced into field soils under a hope to be used as organic fertilizers or under a governmental policy in aim to promote recycling and re-using organic wastes. Some biological wastes were reported to include antibiotic resistant bacteria [3]-[7] which acquired antibiotic resistance during chemo- therapy for human or livestock, or by the addition of antimicrobial growth promoter (AGP). At present, spread- ing of antibiotic resistant bacteria from animal husbandry is becoming a major public health problem, implicat- ing affair of community-acquired infectio n of antibio tic resistant b acteria through undergr ound water, or vegeta- bles, or daily products. Wh ile the existing their surveillan ce method targeting specific nosoco mial bacteria is not suitable for the evaluation of their environmental risk [8] [9]. Because the susceptibility tests and taxonomy de- terminations must be broadly expanded over a large numbers of environmentally important bacterial groups for the risk assessment [10]. If the bacterial taxonomies in culture grown under antibiotics can be elucidated directly without isolation, an- tibiotic resistant b acter ial flora in v arious env ironments will b eco me more eas ily analyz ed , which w ill con tribute to establish their environmental risk assessment method. PCR-RFLP of the 16S rDNA seemed suitable for this purpose, because not only taxonomic estimation of isolates [1] [10] [11], but also that of the mixed culture without isolation [12] can be estimated by measuring number and the lengths (bp) of the restriction fragments. In this manuscript, bacterial taxonomies in culture grown under polymyxin B were determined directly without isolation by employing PCR-RFLP. Furthermore, numbers of each taxonomically different bacterial group in field soils were roughly estimated by the most probable number method after taxonomic elucidations of the mixed culture in the serially diluted culture medium. 2. Materials and Methods 2.1. Enumeration by Dilution Plate Method and MPN Surface soils (depth 0 - 15 cm) were obtained from upland Andosol fields (B field) at Koshi, Kumamoto, Japan (National Agricultural Research Center for Kyushu-Okinawa Region), where buckwheat have been cultivated without organic fertilizer or manure, and from the surface horizon of Gray Lowland paddy field soil (P field) at Koshi, Kumamoto, Japan, where a rice-wheat rotation under paddy-upland fields conditions with an annual ap- plication of chemical fertilizer and organic manure. Sampling was done in duplicate on October 5. Immediately after sampling, soils were sieved (<2 mm). The number of polymyxin B resistant bacteria was estimated by the dilution plate method using the peptone- polymyxin medium (PP medium); 10 g proteose peptone (Difco, Sparks MD), 5 g NaCl, 15 g agar and 5 mg polymyxin B sulfate per liter, pH 7.0 [13] [14]. After 6 days incubation at 30˚C, colony forming units (CFU) wer e coun te d. For MPN, serial 10-fold dilutions (10−2 to 10−7) prepared from soil (1 g fresh wt.) were inoculated to test vials (5 replicates) including PP medium. After 3 days incubation at 30˚C, bacterial DNA in each vial was extracted as the followings. Each bacterial group was counted by MPN after the phylogenetic estimations. 2.2. MERFLP of the Amplified 16S rDNA Chromosomal DNAs of each MPN vials were prepared as described previously and purified by conventional methods. Amplification of 16S rDNA was according to the former studies [1] [10] using the V2 forward primer (41f; 5’GCTCAGATTGAACGCTGGCG3’, corresponding to 22 - 41 positions of 16S rRNA gene of E. col i [15], and the V6 reverse primer (1066r; 3’GTCGAGCACAACACTTTACA5’ corresponding to the 1066 - 1085 positions) [16] [17]. PCR product (10 μl) was separately digested by each of 10 units of the restriction enzyme, HaeIII or HhaI or RsaI or AluI (Takara Bio Co. Ltd. Shiga Japan) in Low salt buffer solution (10x Low salt buf- fer, Takara Bio Co. Ltd.). 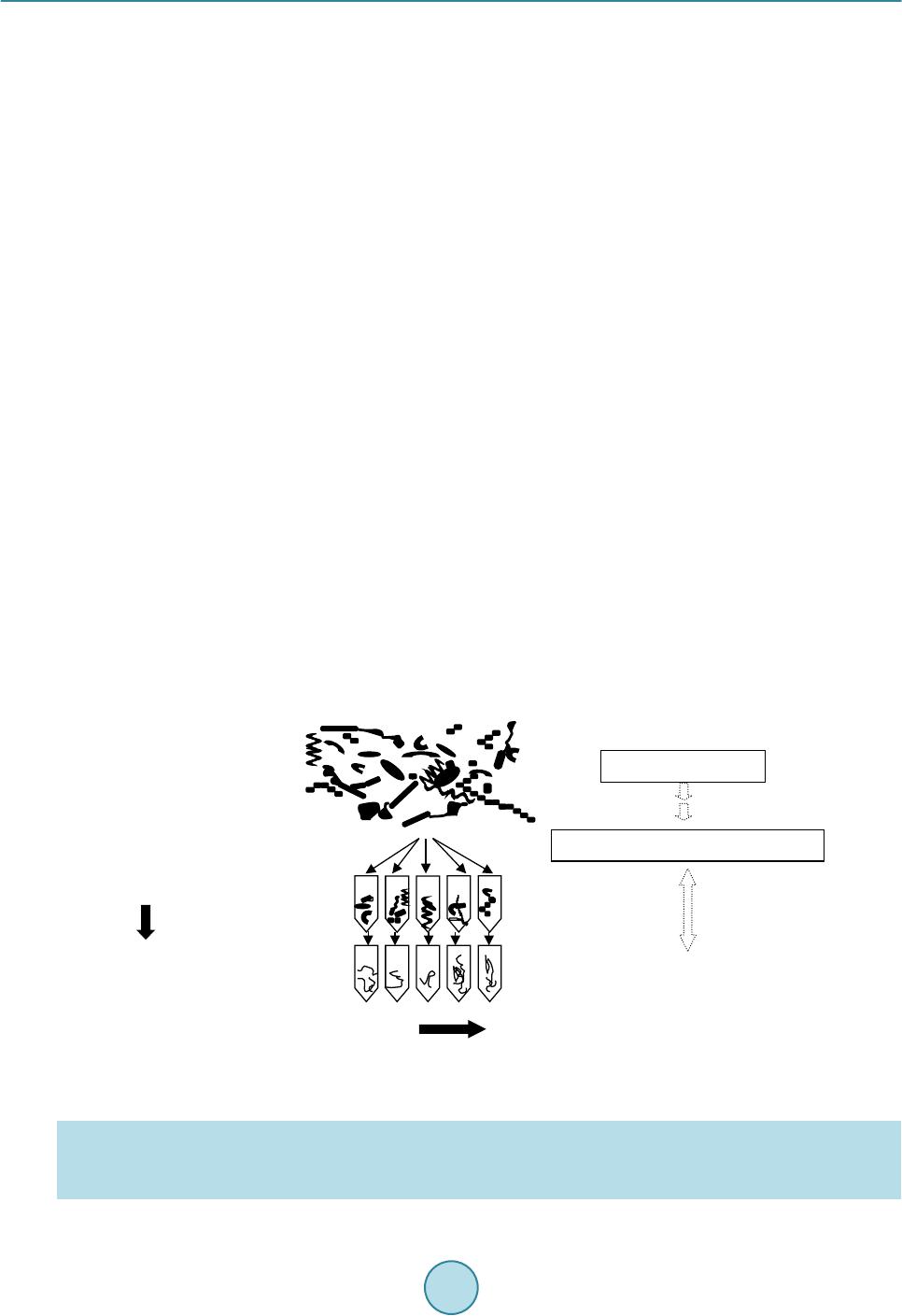 K. Watanabe et al. 2.3. Fragment Lengths Measurement by Microchip Electrophoresis System Fragment lengths was measured by microchip electrophoresis system (Cos mo -i SV1200; Hitachi Electronics Engineering Co., Ltd. Tokyo Japan) as described previously [10] [11]. The sample was diluted by de-ionized water (10 folds for Low salt buff er) before loading on i-tip DNA (IC-1000, Hitach i Chemical Industry Co., Ltd., Tokyo Japan). DNA fragment (65 bp; 5’GCTCAGATTGAACGCTGGCGACATTTCACAACACGAGCTGGCT CAGATTGAACGTGGCGACATT 3’) was used as the lower internal standard, and the PCR product amplified by 41f/1066r primers was used as the upper internal standard, which were co-applied with samples as described previously [10] [11]. In the next similarity search process, the fragment smaller than 100 bp were eliminated from the both theoretical and measured MERFLs as described previously [1]. 2.4. Theoretical Multiple Enzyme Restriction Fragment Lengths (MERFL) Database Used for the Estimation The same theoretical MERFL database as that described previously [1] [10] [11] was used for this research, which was consisted from 4370 sequence files of 576 bacterial genera, and 143 uncultured and 34 unidentified bacteria. 2.5. Data Processing for Phylogenetic Estimation Using Multi-Template DNA and Phylogenetic Estimation (Figure 1) As each MPN vial included mul t i-template DNA originated from heterogeneous bacteria, most of the measured MERFLP was the mixed MERFLPs digested from the heterogeneous 16S rDNA. Whereas all the theoretical MERFLs were originated from the homogeneous 16S rDNA sequence, the measured MERFLP digested from the homoge neous 16S r D NA must be use d for the phylogenetic estimation. In order to search and select the MERFLP digested from the homogeneous 16S rDNA among the mixed MERFLPs, the authors constructed the following procedures: 1) the restriction fragments (RF s) with the highest relative mole concentration (ratio of fluorescent intensity to fragment size) were selected until accumulated RF sizes did not exceed to the original 16S r DNA size (1070 bp ± measuring error), which were used as the major RFs (rep resente d as H in Ta ble 1). 2) After subtraction of the above the major RFs from the mixed heterogene- ous RFs, RFs originated from the 2nd major gene were similarly selected among the remained heterogeneous RFs, and used for similarity search (represented as M in Table 1). 3) After subtraction of the above 2nd major Figure 1. Scheme to identify and quantify each bacterial group using MERFLP and MPN. Theoretical MERFL database was constructed from DNA sequence files by the method of Watanabe and Okuda [2] as described in the former papers [1] [10] [11]. Similarity between the measured MERFLP and the theoretical MERFLP were searched based on pairwise distance ac- cording to Nei and Li [18]. I.MPN 1)Dilution of sample 2)Selective incubation 3)Extraction of DNA II.PCR/MERFLP 1) PCR amplification of 16S rDNA 2) Digestion by restriction enzym e 3) Fragment lengths m easurement by microchip electrophoresis system measurement III. Data processing(Similarity sea rch) 1) Selection of R Fs originated from a sing le gene 2) Phylog ene ti c es ti matio n by the metho d ofWatanabe and Okuda [2] 3) Calculation of each bacterial number by M PN electrophoresis syst em measurement Theoretical MERFL database DNA sequence files  K. Watanabe et al. Table 1. Affiliation of bacteria grown in serially diluted PP medium by MERFLPa. Vial No.b Relative mole ratioc Restriction enzymesd Similarity (%)/ Allowance limite Name (Accession number)f A P10−42H P10−51H P10−53H P10−54H P10−62H P10−63H P10−64H P10−65H B10−43H B10−45H B10−51H B10−54H B10−65H −7 100 100 100 100 100 100 100 100 100 100 100 100 100 Ha, Hh, R, A Ha, Hh, R, A Ha, Hh, R, A Ha, Hh, R, A Ha, Hh, R, A Ha, Hh, R, A Ha, Hh, R, A Ha, Hh, R, A Ha, Hh, R, A Ha, Hh, R, A Ha, Hh, R, A Ha, Hh, R, A Ha, Hh, R, A 100 100 100 100 100 100 100 100 100 100 100 100 100 10 10 10 10 10 10 10 10 10 10 10 10 10 Bacillus cereus (X55060, X55063, B. cereus1, B. cereus2), B. medusa (X60628, B. medusa), B. thuringiensis (X55062, B. thuringi), B. mycoides (B. mycoides, X55061) B P10−43M 35.5 R,A 100 14 Bacillus sp.(B.spAk1) B10−41H 100 Ha, Hh, R, A 85.7 10 B. cycloheptanicus (X51928) B10−44H B10−64H 100 100 Ha, Hh, Ha, Hh 100 100 10 10 B. licheniformis (X60623, B. lichenif), Paenibacillus azotofixans (X60608, Pae.azifi2), P. alvei (Pae.alvei,Pae.alvei3), P. kobensis (Pae.kobens) P10−64M 48.0 Hh, A 100 10 B. pumilus (X60637, B. pumilus) C B10−65L 1.0 Ha, A 100 15 Clostridium malenominatum (C. malenomi) P10−43H 100 R, A 100 10 C. histolyticum (M59094, C. histolyt), C. collagenovorans (C. colgenovo), Micromonospora chalcea (D85489) P10 4M B10−65M 2.9 3.5 Ha, Hh, A Ha, Ag 100 90.0 14 10 C. tyrobutyricum (M59113), C. tyrobutyricum (C. tyrobu51, C. tyrobuty)g D P10−53M B10−74M 5.0 3.2 Ha, A Ha, R, A 100 64 15 10 Lactobacillus mali (M58824, L. mali), L. hilgardii (M58821, L. hilgardi) B10−44M 6.4 Hh, A 100 10 Heliobacterium fasciatum (L36197) B10−51M 13.0 R. A 100 11 Erysipelothrix rhusiopathiae (Ers. rhusio, M23728) P10−41L 30.5 Ha, Hh 100 11 Mycoplasma sualvi (M. sualvi) P10−53L 3.0 Ha, A 90.0 10 Pediococcus urinaeequi (D87667) P10−63M 4.4 Ha, Hh, R, A 68.3 10 Simkania negevensis (Smk. negevn) P10−43L 10.5 Hh, R 100 10 Spiroplasma mirum (M24662), S. (Spp.cit2HP), S. poulsonii (Spp.poulsn) B10−64M 2.5 Hh, A 100 15 Haloanaerobacter chitinovoran (U32596) E B10−52M 4.5 Ha, Hh, R, A 66 10 Actinokineospora riparia (AF114802) B10−44L 3.5 Ha, R, A 66 10 Clavibacter xyli (M60935) F P10−41M 54.5 Ha, R 100 13 Holospora obtusa (X58198) B10−54M 22.5 Ha, Hh, R, A 69.3 10 Burkholderia glathei (AB021374, Bur. glath1) B10−52H 100 Ha, Hh, R, A 100 10 Ralstonia pickettii (Ral. picke5) B10−43M 1.0 Ha, R 87.5 10 Oxabacter pfennigii (Ox. pfennig) P10−42L 4.7 Ha, Hh, A 65.2 10 Pseudomonas fragi (D84014), P. stutzeri (U25432, U26262, D84024), P. citronellolis (Z76659) P10−55H 100 Ha, Hh, A 100 14 Vibrio diazotrophicus (V. diazotro) B10−51L 0.9 Ha, R 83.3 10 Pelobacter propionicus (Peb. propio)  K. Watanabe et al. Continued G P10−64L P10−62M 3.9 3.7 Hh, R, A Hh, R, A 82.2 72.2 10 10 Prevotella numinicola (AB003401) P10−65M P10−54L 4.3 8.3 R, A Ha, Hh, R, A 87.5 68.3 10 10 Prevotella oralis (L16480) H B10−45M P10−51M 13.0 8.7 R, A Ha, Hh, A 100 68.9 10 10 Flexibacter litoralis (Flx. litora, M58784) P10−62L 0.8 R,A 83.3 10 Flexibacter maritimus (Flx. marit2) P10−42M 42.3 Ha, Hh, A 100 13 Cryseobacterim balustinum (M58771), Flabovacterium indoltheticum (M58774) P10−51L 0.9 Ha, A 51 10 Taxeobacter ocellatus (Y18835) I P10−63L 0.7 Ha, R, A 64.4 10 Oscillatoria agardhii (X84811) P10−55M 12.5 Ha, A 90 10 Fervidobacterium islandicum (M59176) aGrouping was based on affiliation by MERFL; Bacillus cereus group (Group A), the other Bacillus spp.(Group B), Clostridium spp. (Group C), the other Firmicutes (Group D) , Actinobacteri a (Group E), Proteobacteria (Group F), Prevotella spp. (Group G) , the other Cyto phagales (G roup H), and the other gram negative bacterial group (Group I). bThe 1st letter in vial indicates soils; “P” stands for paddy filed soil, and “B” stands for up l an d field soil. Exponenti al of vial number r ep r es ents the deci mal dilu tion o f t h e vi al . The 2nd number of vial number (1-5) represents number i n 5 replicates f or the each decimal dilution. H of last letter represents MERFL originating from the major 16S rDNA, M represents from the 2nd major 16S rDNA, and L represent s f rom the 3rd major 16S rDNA. cRelative mole ratio was a ratio of averaged relative mole concentrations of the 2nd, or 3rd major MERFL to those of the major MERFL as described in th e materials and meth od. dRestricti on enzymes used f or similarity sear ch; “Ha”, “Hh”, “R”, and “A” stand for Hae III, Hha I, Rsa I, and Alu I. eAllowance li mit for measur ing error was s et at 10% at first, then i ncreased as descr ibed in mat erials and methods. For the measured MERFLP which had no completely identical theoretical MERFLP, the theoretical MERFLP having the highest similarity using all the RFLPs was presented with the similarity in italics as described in the materials and method. fSpecies name (accession number) of the theoretical MERFL having the highest similarity with the measured MERFL of the isolate. gAdditional name (accession number) of the theoretical MERFL ha vi ng t he same MERFL for the used restriction enzymes. RFs from the remained heterogeneous RFs, RFs originated from the 3rd major gene were similarly selected and used for similarity search (represented as L in Table 1). The similarity between the measured RFLP (A) and the theoretical RFLP (B) was calculated as described previously [1] [10] [11] based on the pairwise distance (DAB) by the following equation; DAB = 1 − 2NAB/(NA + NB), where NA and NB were the numbers of fragments of each RFLPs and NAB was the number of shared frag- ments that indicated same sizes within an allowance limit for measuring error according to Nei and Li [18]. The pairwise distance of the MERFLPs (DABME) was an average of all the DABs for used restriction enzymes. S imilar- ity (%) was (1 − DABME) × 100 (Table 1). In similarity search, allowance limit for measuring error was set at 10% in the first analysis, then gradually increased to 15 % until completely iden tical theoretical MERFL (100 % similarity) were found out (Table 1) [1] [10] [11]. If the completely identical theoretical MERFL was not found out by using all of the measured MERFL data, combinations of restriction enzymes used for the analysis was changed (Table 1) [1] [10] [11]. As to the measured MERFL which had no completely identical the oretical MERFL, the theoretica l MERFL having the highest similarity to the measured MERFL was indicated in Table 1 [1] [10] [11]. 2.6. Estimation of Numbers of Each Taxonomically Different Group by the Most Probable Number Method Most probable numbers of each groups (A-I) were estimated for five-tube, three-decimal-dilution experiment (Table 2). Confidence limits sh own in Table 2 were obta ined usin g Woodwar d’s method [19]. 3. Results and Discussion 3.1. Enumeration by Dilution Plate Method Bacterial counts on PP medium in the both fields (P field, 1.82 ± 0.65 × 106 CFU∙g−1 dry soil; B field, 2.30 ± 1.03 × 106 CFU∙g−1) were similar level to those in uncultivated soil (0.2 × 106 CFU∙g−1) [14], and cultivated upland soil(1.2 × 106 CFU∙g−1) [14], and smaller than that in the fallow land area (from 9.2 to 18.7 × 106 CFU g−1) [20], and much s maller than that in the field soil applied with 120 t liquid livestock feces (from 49.3 to 72.1 × 106 CFU∙g−1 dry soil) [20] and that with 600 t field (84.1 to 64 5 × 106 CFU∙g−1) [20].  K. Watanabe et al. Table 2. Most probable numbers of each groups (A - I) and 5% confidence limits obtained using Woodward’s method [19]. Groups Paddy field soil Upland field soil Three dilutions Score MPN∙g−1 dry soil 5% limits Three dilutions Score MPN∙g−1 dry soil 5% limits Low High Low High A. B. cereus 10−5 10−6 10−7 3-4-0 33.01 - - 10−5 10−6 10−7 2-1-1 13.24 4.31 34.53 B. Bacillus spp. 10−5 10−6 10−7 0-1-0 2.83 1.57 15.72 10−5 10−6 10−7 0-1-0 2.59 1.44 14.44 C. Clostridium 10−4 10−5 10−6 1-1-0 0.63 0.15 2.36 10−5 10−6 10−7 0-2-0 5.323 1.44 18.71 D. The other Firmicutes 10−5 10−6 10−7 2-1-0 10.69 3.14 33.02 10−5 10−6 10−7 1-1-1 8.78 2.88 25.90 E. Actinobacteria 10−4 10−5 10−6 1-1-0 0.58 0.14 2.16 F. Proteobacteria 10−4 10−5 10−6 2-1-0 1.069 0.31 3.30 10−4 10−5 10−6 3-1-0 1.58 0.58 4.17 G. Prevotella 10−4 10−5 10−6 1-3-0 13.05 - - H. Cytophagales 10−5 10−6 10−7 2-1-0 10.6 9 3.14 33.02 10−4 10−5 10−6 1-0-0 0.29 0.14 1.58 I. Gram negative bacteria 10−5 10−6 10−7 1-1-0 6.29 1.57 23.58 3.2. Affiliation of Bacteria by MERFLP without Isolation Affiliations of fifty th ree MERFLPs in each MPN vials were summarized in Table 1. Th ey were affiliated to be Bacillus cereus group (Group A, 15 MERFLPs), the other Bacillus spp. (Group B, 5 MERFLPs), Clostridium spp. (Group C, 4 MERFLPs), the other Firmicutes (Group D, 9 MERFLPs), Actinobacteria (Group E, 2 MERFLPs), Proteobacteria (Group F, 7 MERFLPs), Prevotella spp. (Group G, 4 MERFLPs), the other Cyto- phagales (Group H, 5 MERFLPs), and the other gram negative bacterial group (Group I, 2 MERFLPs). Our former results indicated th at most of the bacterial 16S rDNA were a mplified by using the same PCR condition, and affiliated to the corresponding bacteria. When the method was used for the mixed culture, PCR bias was found to effect on the results as the followings. The most of the major MERFL (21 vials) in the dilution vials were affiliated to be B. cereus (15 vials), while there was no vials in the 2nd major and 3rd MERFL affiliated to be B. cereus (Table 1 and Table 3). The results suggested that B. cereus had always been the major MERFL even if B. cereus existed as the minor bacteria in the vial, because 16S rDNA of B. cereus might be amplified preferentially to th e other b a cterial groups by the used PCR condition due to the PCR bias. Although the MERFLP from the homogeneous 16S rDNA, the major MERFL, the 2nd major MERFL, and the 3rd major MERFL, could be selected among the mixed MERFLPs, the major MERLP provided the most re- liable information on bacterial taxonomy of each bacterial group by the similarity search as the followings. Ratio of the MERFLs having 100% similarity to the corresponding theoretical MERFLs was the highest in the major MERFL (90.5%), followed by the 2nd major MERFL (50.0%), and the 3rd major MERFL (25.0%) (Table 3), which indicated that the discrepancies between the measured MERFL and the corresponding theoretical MERFL was gradually increasing in the 2nd and 3rd major MERFL. Smaller peak area of 2nd major MERFL (14.5% of the major MERFL Table 3) and the 3rd major MERFL (5.73% of the major MERFL Table 3) might be one factor to increase the discrepancies caused by the undetectable fragment under the detection limit or misreading of the fragment size. The 2nd major, or the 3rd major MERFLP having the same fragment size as the major MERFLP might be another factor to increase the discrepancies. Because the fragment, which was once attri- buted to the major MERFLP, could not be attributed to the 2nd major, or the 3rd major MERFLP. 3.3. Enumeration of Each Bacterial Group by MPN Bacillus cereus group (Group A) w as estimated to be 3.30 × 106 MPN∙g−1 dry soil in P field, and 1.32 × 106 MPN∙g−1 in B filed (Tab le 2 and Figure 2). The other Bacillus spp. (Group B) was enumerated to be 0.28 × 106 MPN∙g−1 in P field, and 0.26 × 10 6 MPN∙g−1 in B field (Table 2 and Figure 2). Clostrid ium spp. (Group C) was enumerated to be 0.06 × 106 MPN∙g−1 in P field, and 0.53 × 106 MPN∙g−1 in B field (Table 2 and Figure 2). The other Firmicutes (Group D) were enumerated to be 1.07 × 106 M PN∙g−1 in P field and 0.88 × 106 MPN∙g−1 in B 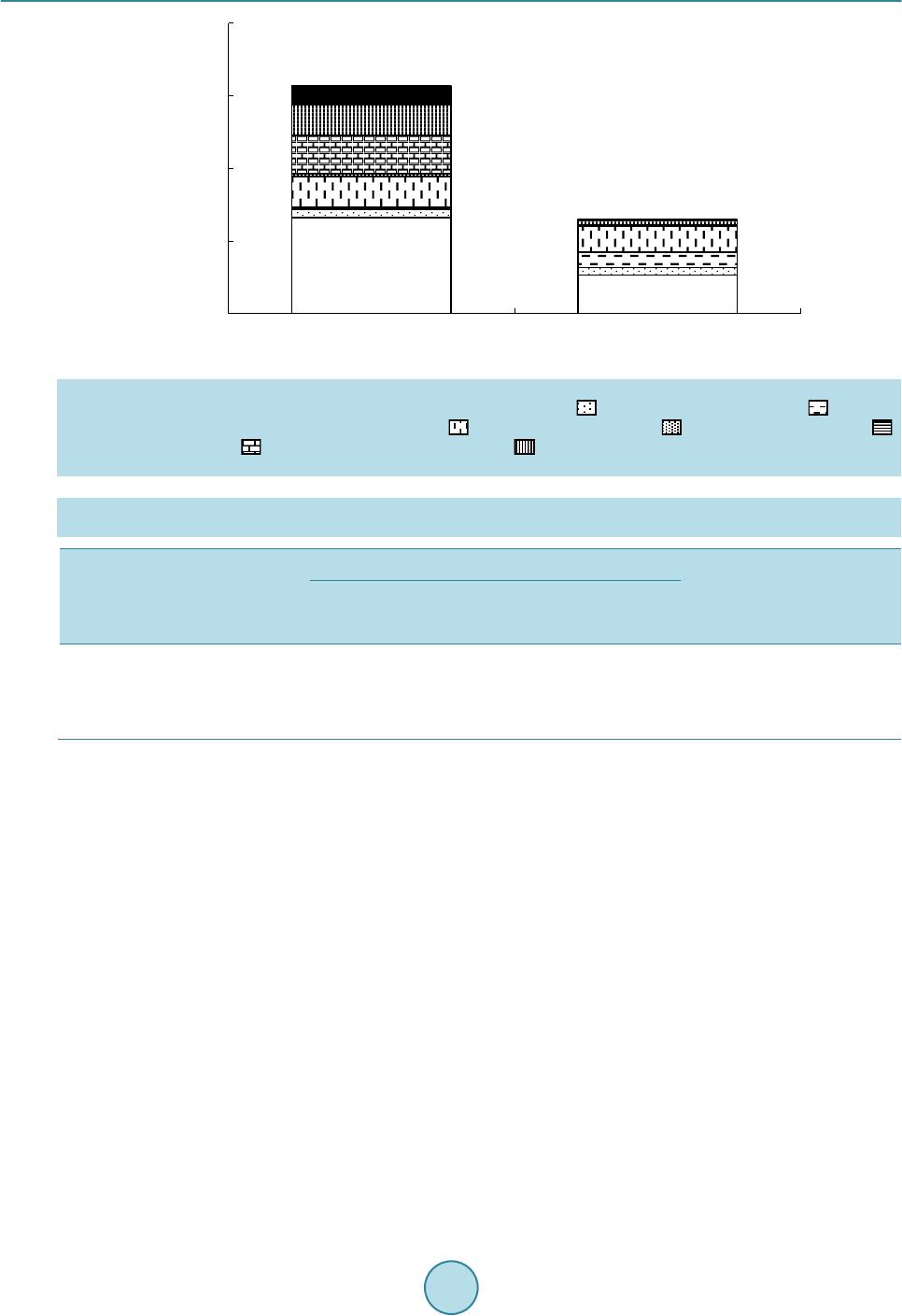 K. Watanabe et al. Figure 2. Numbers of bacterial groups estimated by MPN and MERFLP in paddy field soil (P), and upland field soil (B). Number of B. cereus group (Group A; □), the other Bacillus spp. (Group B; ), Clostridium spp., (Group C; ), the other low GC content gram positive bacterial group (Group D; ), Actinobacteria (Group E; ), Proteobacteria (Group F; ), Prevotella spp. (Group G; ), the other Cytophagales (Group H; ), and the other bacterial group (Group I; ■) were pre- sented. Table 3. Affiliated bacterial groups of the major, the 2nd major, and 3rd major MERFLs and their relation to the results of similarity search. Bacterial groups Ratio of the vails with 100% similarity Relative mole concentration* B. cereus group (A; 15 vials) Gram positive bacteria (B - E; 20 vails) Gram negative bacteria (F - I; 18 vails) The major MERFL (21 vials) 15 4 2 90.5% 100% The 2nd major MERFL (20 vials) 0 11 9 50.0% 14.5% The 3rd major MERFL (12 vials) 0 5 7 25.0% 5.73% *A ratio of averaged relative mole concentration s of the 2nd, or 3rd major MERFL to those of the major MERFL. field (Table 2 and Figure 2). Proteobacteria (Group F) were enumerated to be 0.10 × 106 MPN in P field and 0.16 × 106 MPN in B field (Tabl e 2 and Figure 2). Prevotella spp.(Group G), the o ther Cytop hagales (G roup H), and the other bacterial group (Group I) were enumerated to be 1.31 × 106 MPN, 1.07 × 106 MPN, and 0.63 × 10 6 MPN∙g−1 in P field, while the other Cytophagales (Group H) was enumerated to 0.028 × 106 MPN∙g−1 in B field (Table 2 and Figure 2). Most MPN scores remained low especially in lower decimal dilution except for the some groups, Group A in P field and Group F in B field ( Table 2 ), which might be caused by the PCR bias as the followings. When spe- cific 16S rDNA, e.g., B cereus group, was preferentially amplified, the other 16S rDNAs in the same vials could not be equally amplified, which might inhibit reliable taxonomic estimation even if they were numerically do- minant genomic DNA. The PCR bias also seemed to cause the inappropriate MPN scores (3-4-0, 1-3-0 Table 2) in some bacterial grou ps (Group A and Group G in P field) (Table 2), where confidence limit could not be cal- culated. Inhib ition of P CR in low er dilution v ials, which was caused by inco mplete removal of PCR inhibitor in soils, also caused the lower MPN score in the lower decimal dilution. Although the numbers of some bacterial groups might be underestimated due to the PCR bias and PCR inhibition described above, the method at least provided the minimum numbers of each bacterial group. 4. Conclusions Polymyxins (B and E), polycationic peptide antibiotics produced by Bacillus polymyxa , were bactericidal to  K. Watanabe et al. gram-negative bacteria (MIC50 = 1 ppm for most gram-negative bacteria) except for Burkholderia cepacia [21] due to crossing the bacterial outer membrane by competitive divalent cation displacement by bulky polycations and little to no effect on gram-positive bacteria. The bacteria in MPN vials, which were affiliated to be gram negative bacteria (from Group F to Group I, Figure 2), were suggested to acquire antibiotic resistance [10] [22]. Although polymyxin B have mainly been used in hospitals and have never been used as AGP, or applied for li- vestock, polymyxin E (colistin), which h as a similar structure and has the same site of action, has been used as an AGP in Japan. Polymyxin B-resistant gram negative bacteria detected in this study were supposed to be co- listin resistant bacteria, because large numbers of polymyxin B (over 106 CFU/g dry matter) and colistin-resis- tant bacteria (over 106 MPN/g dry matter) were typically found in raw livestoc k feces [23]. The estimated numbers of gram negative antibiotic resistant bacteria (3.11 × 106 MPN∙g−1 in P, and 0.19 × 106 MPN∙g−1 in B) were lower than those estimated for the other field soils (from 31.7 × 106 CFU∙g−1 to 258 × 106 CFU∙g−1) [10] [20] where large amount of liquid livestock feces had annually been applied. As considerable numbers of Prevotella spp., and the other Cytophagales (1.31 × 106 MPN, 1.07 × 106 MPN), which had been suggested to be of feces origin [ 10] [24] were only enumerated in P field, the gram negative antibiotic res istant bacteria were concluded to be contaminated from organic manure. The method presented here was found to be effective and useful for the purpose of monitoring whole antibi- otic resistant bacterial flora in the environment and evaluating their risk. As the method was a new method, which was different from the other known analysis methods for microbial group such as denaturing gradient gel electrophoresis (DGGE) [25] [26] or terminal restrictio n fragment length polymorphism (t-RFLP) [27] [28], the availability as evaluation method for the other microbial groups, such as multi-drug resistant bacteria, bacteria causing food poisoning, and the other general bacteria, a precision of the affiliation, and validation of enumera- tion of each microbial group will be described in the next manuscripts. Acknowledgements The most of this work had been achieved in Research Team for Biomass Recycling System, National Agricul- tural Research Center for Kyushu-Okinawa Region. The author thanks Prof. H. Tamura, and Dr. A. Hosoda, Meijyo University, Dr. H. Yosikawa, the former Prof. of Fukuoka Institu te of Technology, for their suggestion s and encouragements during this work. References [1] Watanabe, K., Okuda, M. and Koga, N. (2008) A Newly Developed System Based on Multiple Enzyme Restriction Fragment Length Polymorphism—An Application to Proteolytic Bacterial Flora Analysis. Soil Science and Plant Nu- trition, 54, 204-215. http://dx.doi.org/10.1111/j.1747-0765.2007.00230.x [2] Watanabe, K. and Okuda, M. (2003) Method and System for Searching for Relationships between Base Sequences in genes. Japanese Patent No. 3431135, US Patent No. 7006924. [3] Burgos, J.M., Ellington, B.A. and Varela, M.F. (2005) Presence of Multidrug Resistant Enteric Bacteria in Dairy Farm Topsoil. Journal of Dairy Science, 88, 1391-1398. http://dx.doi.org/10.3168/jds.S0022-0302(05)72806-X [4] Nhung, N.T., Cuong, N.V., Campbell, J., Hoa, N.T., Bryant, J.E., Truc, V. N.T ., Kiet, B.T., Jombart, T., Trung, N.V., Hien, V.B., Thwaites, G., Baker, S. and Carrique-Mas, J. (2015) High Levels of Antimicrobial Resistance among Escherichia coli Isolates from Livestock Farms and Synanthropic Rats and Shrews in the Mekong Delta of Vietnam. Applied and Environmental Microbiology, 81, 812-820. http://dx.doi.org/10.1128/AEM.03366-14 [5] Witte, W. (1998) Medical Consequences of Antibiotic Use in Agriculture. Science, 279, 996-997. http://dx.doi.org/10.1126/science.279.5353.996 [6] Sawant, A.A., Hegde, N. V. , Straley, B.A. , Donaldson, S.C., Love, B.C., Knabel, S.J. and Jayarao, B.M. (2007) An ti- microbial-Resistant Enteric Bacteria from Dairy Cattle. Applied and Environmental Microbiology, 73, 156-163. http://dx.doi.org/10.1128/AEM.01551-06 [7] Bradford, P.A., Petersen, P.J., Fingerman, I.M. and White, D.G. (1999) Characterization of Expanded-Spectrum Ce- phalosporin Resistance in E. coli Isolates Associated with Bovine Calf Diarrhoeal Disease. Journal of Antimicrobial Chemotherapy, 44, 607-610. http://dx.doi.org/10.1093/jac/44.5.607 [8] Rahube, T.O., Marti, R., Scott, A., Tien, Y.-C. , Murray, R., Sabourin, L., Zhang, Y., Duenk, P., Lapen, D.R. and Topp, E. (2014) Impact of Fertilizing with Raw or Anaerobically Digested Sewage Sludge on the Abundance of Antibiot- ic-Resistant Coliforms, Antibiotic Resistance Genes, and Pathogenic Bacteria in Soil and on Vegetables at Harvest.  K. Watanabe et al. Applied and Environmental Microbiology, 80, 6898-6907. http://dx.doi.org/10.1128/AEM.02389-14 [9] Marti, R., Tien, Y.-C., Murray, R., Scott, A., Sabourin, L. and Topp, E. (2014) Safely Coupling Livestock and Crop Production Systems: How Rapidly Do Antibiotic Resistance Genes Dissipate in Soil following a Commercial Applica- tion of Swine or Dairy Manure? Applied and Environmental Microbiology, 80, 3258-3265. http://dx.doi.org/10.1128/AEM.00231-14 [10] Watanabe, K. (2008) Application of Multiple Enzyme Restriction Fragment Length Polymorphism Analysis and Mi- crochip Electrophoresis for Estimation of Antibiotic-Tolerant Bacterial Group. Journal of Pesticide Science, 33, 249-260. http://dx.doi.org/10.1584/jpestics.G08-04 [11] Watanabe, K. and Koga, N. (2009) Use of a Microchip Electrophoresis System for Estimation of Bacterial Phylogeny and Analysis of NO3− Reducing Bacterial Flora in Field Soils. Bioscience Biotechnology and Biochemistry, 73, 479- 488. http://dx.doi.org/10.1271/bbb.70712 [12] Chakraborty, S., Persaud, V., Vanegas, S., Gautier, G. and Esiobu, N. (2014) Analysis of the Human Oral Microbiome of Smokers and Non-Smokers Using PCR-RFLP and Ribotyping. Advance in Microbiology, 4, 681-691. http://dx.doi.org/10.4236/aim.2014.410073 [13] Akiba, Y. and Katoh, K. (1986) Microbial Ecology of Bacillus thuringiensis V. Selective Medium for Bacillus thuri n- giensis Vegetativ e Cells. Applied and Entomological Zoology, 21, 210-215. [14] Watanabe, K. and Hayano, K. (1995) Seasonal Variation of Soil Protease Activities and Their Relation to Proteolytic Bacteria and Bacillus Spp in Paddy Field Soil. Soil Biology & Biochemistry, 27, 197 -203. http://dx.doi.org/10.1016/0038-0717(94)00153-R [15] Brosius, J., Palmer, M.L., Kennedy, P.J. and Noller, H.F. (1978) Complete Nucleotide Sequence of a 16S Ribosomal RNA Gene from Escherichia coli. Proceedings of the National Academy of Sciences of the United States of America, 75, 4801-4805. http://dx.doi.org/10.1073/pnas.75.10.4801 [16] Schewieger, F. and Tebbe, C.C. (1998) A New Approach To Utilize PCR-Single-Strand-Conformation Polymorphism for 16S rRNA Gene-Based Microbial Community Analysis. Applied and Environmental Microbiology, 64, 4870-4876. [17] Weidner, S., Arnold, W. and Puhler, A. (1996) Diversity of Uncultured Microorganisms Associated with the Seagrass Halophila stipulacea Estimated by Restriction Fragment Length Polymorphism Analysis of PCR-Amplified 16S rRNA Genes. Applied and Environmental Microbiology, 62, 766-771. [18] Nei, M., and Li, W. H. (1979) Mathematical Model for Studying Genetic Variation in Terms of Restriction Endonuc- leases. Proceedings of the National Academy of Sciences of the United States of America, 76, 5269-5273. http://dx.doi.org/10.1073/pnas.76.10.5269 [19] Haas, C.N. (1989) Estimation of Microbial Densities from Dilution Count Experiments. Applied and Environmental Microbiology, 55, 1934-1942. [20] Watanabe, K. and Niimi, H. (2005) Changes in Soil Protease Activity and Numbers of Culturable Bacteria in Upland Fields by the Application of Liquid Livestock Feces. Soil Science and Plant Nutrition, 51, 491-496. http://dx.doi.org/10.1111/j.1747-0765.2005.tb00057.x [21] Gales, A.C. Reis, A.O. and Jones, R.N. (2001) Contemporary Assessment of Antimicrobial Susceptibility Testing Me- thods for Polymyxin B and Colistin: Review of Available Interpretative Criteria and Quality Control Guidelines. Journal of Clinical Microbiology, 39, 183-190. http://dx.doi.org/10.1128/JCM.39.1.183-190.2001 [22] Tam, V.H., Schillin, A.N., Vo, G., Kabbara, S., Kwa, A.L., Wiederhold, N.P. and Lewis, R.E. (2005) Pharmacody- namics of Polymyxin B against Pseudomonas aeruginosa. Antimicrobial Agents and Chemotherapy, 49, 3624-3630. http://dx.doi.org/10.1128/AAC.49.9.3624-3630.2005 [23] Watanabe, K., Tanaka, A., Yakushido, K. and Tamura, H. (2007) Evaluation of Antibiotic Resistant Bacteria in Or- ganic Wastes by Newly Developed Method for Identification and Quantification of Microbial Ecology. 32th Annual Conference of Pesticide Science Society of Japan, Tokyo, April 2007, 65. [24] Fogarty, L.R. and Voytek, M.A. (2005) Comparison of Bacteroides-Prevotella 16S rRNA Genetic Markers for Fecal Samples from Different Animal Species. Applied and Environmental Microbiology, 71, 5999-6007. http://dx.doi.org/10.1128/AEM.71.10.5999-6007.2005 [25] Satokati, R.M., Vaughan, E., Akkermans, A.D.L., Saarela, M. and Vos, W.M. (2001) Bifidobacterial Diversity in Hu- man Feces Detected by Genus-Specific PCR and Denaturing Gradient Gel Electrophoresis. Applied and Environmental Microbiology, 67, 504-513. http://dx.doi.org/10.1128/AEM.67.2.504-513.2001 [26] Muyzer, G., Waal, E.C.D. and Uitterlinden, A.G. (1993) Profiling of Complex Microbial Populations by Denaturing Gradient Gel Electrophoresis Analysis of Polymerase Chain Reaction-Amplified Genes Coding for 16S rRNA. Applied and Environmental Microbiology, 59, 695-700. [27] Kent, A.G., Smith, D. J., Benson, B.J. and Triplett, E.W. (2003) Web-Based Phylogenetic Assignment Tool for Analy- sis of Terminal Restriction Fragment Length Polymorphism Profiles of Microbial Communities. Applied and Envi-  K. Watanabe et al. ronmental Microbiology, 69, 6768-6776. http://dx.doi.org/10.1128/AEM.69.11.6768-6776.2003 [28] Marsh, T.L., Saxman, P.L., Cole, J. and Tiedje, J.M. (2000) Terminal Restriction Fragment Length Polymorphism Analysis Program, a Web-Based Research Tool for Microbial Community Analysis. Applied and Environmental Mi- crobiology, 66, 3616-3620. http://dx.doi.org/10.1128/AEM.66.8.3616-3620.2000
|