Journal of Biomedical Science and Engineering
Vol.6 No.1(2013), Article ID:27082,9 pages DOI:10.4236/jbise.2013.61006
Favorable and unfavorable amino acid residues in water-soluble and transmembrane proteins
![]()
Department of Clinical Laboratory Science, Graduate Course of Medical Science and Technology, School of Health Science, Kanazawa University, Kanazawa, Japan
Email: naka@kenroku.kanazawa-u.ac.jp, kkitamur@mhs.mp.kanazawa-u.ac.jp
Received 26 November 2012; revised 28 December 2012; accepted 5 January 2013
Keywords: Amino Acid Composition; Nucleotide Composition; Favorable and Unfavorable Residues; Water-Soluble and Transmembrane Proteins; Thermophilic and Mesophilic Species
ABSTRACT
We analyzed the amino acid residues present in the water-soluble and transmembrane proteins of 6 thermophilic and 6 mesophilic species of the domains Archaea and Eubacteria, and characterized them as favorable or unfavorable. The characterization was performed by comparing the observed number of each amino acid residue to the expected number calculated from the percentage of nucleotides present in each gene. Amino acids that were more or less abundant than expected were considered as favorable or unfavorable, respectively. Comparisons of amino acid compositions indicated that the water-soluble proteins were rich in charged residues such as Glu, Asp, Lys, and His, whereas hydrophobic residues such as Trp, Phe, and Leu were abundant in transmembrane proteins. Interestingly, our results found that although the Trp residue was abundant in transmembrane proteins, it was not defined as favorable by our calculations, indicating that increased numbers of a particular amino acid does not necessary indicate it is a favorable residue. Amino acids with high G + C content such as Ala, Gly, and Pro were frequently observed as favorable in species with low G + C content. Comparatively, amino acids with low G + C content such as Phe, Tyr, Lys, Ile, and Met were frequently observed as favorable in species with high G + C content. These are the examples to increase the supply of amino acids than expected. Amino acids with neutral G + C content, i.e., Glu and Asp were favorable in water-soluble proteins from all species analyzed, and Cys was unfavorable both in water-soluble and transmembrane proteins. These results indicate that amino acid compositions are essentially determined by the nucleotide sequence of the genes, and the amino acid content is altered by a deviation from expectation.
1. INTRODUCTION
Proteins can be roughly classified into 2 types: water-soluble proteins and transmembrane proteins. The transmembrane proteins have membrane-spanning regions, which contact the hydrophobic environment of the lipid bilayer and are largely composed of amino acids with nonpolar side chains [1-3]. Comparatively, water-soluble proteins have more charged residues than transmembrane proteins, and therefore, the amino acid compositions differ between the 2 types of proteins. We recently reported that the dinucleotide composition of the genes coding for water-soluble proteins differs from those encoding transmembrane proteins [4]. The genes encoding water-soluble proteins are rich in the purine dimers AA, AG, and GA, whereas those encoding transmembrane proteins are rich in the pyrimidine dimers TT, CT, and TC. This trend was observed in thermophilic and mesophilic species of Archaea and Eubacteria. The AA, AG, and GA dinucleotides are components of the codons of the charged residues, Glu, Asp, Lys, and Arg, whereas the TT, CT, and TC dinucleotides are components of the codons of the hydrophobic residues Leu, Ile, and Phe. The AA, AG, and GA dinucleotides are complementary to TT, CT, and TC, this revealed that a simple strategy is utilized to produce water-soluble and transmembrane proteins with distinct characteristics by using the DNA sequences on opposing strands.
The primary structure of a protein depends on the nucleotide composition of the protein-coding gene. Therefore, if the order of the coding nucleotides is random, the amino acid content would correlate with the calculated values determined by the nucleotide composition. The G + C content of bacterial genomes varies from 25% to 75% between species, but it is relatively constant within a bacterial genome [5,6]. The nucleotide sequences of bacterial genes have species-specific dinucleotide compositions [7-9]. Previous studies identified correlations between the nucleotide composition of genes and the amino acid content of proteins on a genome-wide scale [10-12]. However, as water-soluble and transmembrane proteins have different amino acid and nucleotide compositions, it is necessary to analyze them separately like Lobry’s study [13]. Studies of amino acid compositions from various species have revealed that the proteins of thermophiles have more charged amino acids than the proteins of mesophiles [14-18], whereas halophilic proteins contain more Asp residues [19].
In this study, we analyzed amino acid compositions in water-soluble and transmembrane proteins taking into account of different character of the coding sequences in their nucleotide compositions. We characterized amino acids as favorable or unfavorable depending on whether they were observed more or less often than expected. The favorable and unfavorable residues was used to understand the relationship between G + C content and protein compositions in the thermophilic and mesophilic Archaea and Eubacteria species in a wide range of G + C content.
2. MATERIALS AND METHODS
2.1. Sequence Retrieval
The species surveyed in this study were 3 thermophilic Archaea, Sulfolobus tokodaii [20], Archaeoglobus fulgidus [21], and Methanopyrus kandleri [22], 3 thermophilic Eubacteria, Thermoanaerobacter tengcongensis [23], Thermotoga maritima [24], and Thermus thermophilus HB8 (Genbank: AP008226.1), 3 mesophilic Archaea, Methanosphaera stadtmanae [25], Methanocorpusculum labreanum [26], and Halobacterium sp. NRC-1 [27], and 3 mesophilic Eubacteria, Haemophilus influenzae Rd KW20 [28], Escherichia coli K12 MG1655 [29], and Pseudomonas aeruginosa PA01 [30]. The species were selected arbitrarily, however, we ensured that they covered a wide range of genomic G + C content. Their genome sequences were retrieved from the web FTP site (ftp://ftp.ncbi.gov/genomes/) of the National Center for Biotechnology Information (NCBI). The protein-coding nucleotide sequences and amino acid sequences were retrieved from NCBI as ffn and faa files.
2.2. Selection of Water-Soluble and Transmembrane Proteins
The proteins were classified as water-soluble or transmembrane proteins according to the annotations on the genome to protein structure and function (GTOP) database [31]. The SOSUI program [3] was used in the GTOP database to predict the transmembrane regions. Proteins with no transmembrane regions were considered as water-soluble proteins. Proteins with ≥2 transmembrane regions were utilized to calculate the amino acid composition of transmembrane proteins. The transmembrane proteins were divided into 100 groups and one protein was randomly selected from each group. The water-soluble proteins were similarly selected. The water-soluble and transmembrane proteins were examined for their amino acid sequence similarity by using the BLAST program [32]. Proteins which had ≥30% sequence identity with other selected proteins were replaced to keep the sequence identity below 30%. Amino acid sequences utilized in this study correspond to the genes in our previous study [4]. Proteins were longer than 100 residues.
2.3. Ratios of Observed and Calculated Compositions
The expected amino acid composition was calculated as the product of the mononucleotide content for each gene. For example, a gene consisting of 31.4% adenine, 20.0% cytosine, 26.4% guanine, and 22.2% uracil would have an expected frequency of Lys residue (AAA and AAG) of 0.314 × 0.314 × 0.314 + 0.314 × 0.314 × 0.264 = 0.0570. The 3 stop codons were not included in the calculation of expected values, therefore, the values were adjusted by a correction factor of 1.062. Thus, in this example, the expected frequency of Lys residues was 6.05%. The expected amino acid compositions for 100 water-soluble and transmembrane proteins were calculated and averaged. These values were then compared to the average observed number, and the ratios of the observed values to the expected values were calculated.
The expected dinucleotide composition was calculated as the product of the mononucleotide composition for each gene. The averages of the expected dinucleotide compositions for 100 genes encoding water-soluble and transmembrane proteins were calculated. Subsequently, the ratios of the observed values to the expected dinucleotide composition were calculated.
3. RESULTS
3.1. Amino Acid Composition
The average amino acid compositions of 100 water-soluble and 100 transmembrane proteins from 12 species are listed in Table 1 with the G + C content of their genes. In T. maritima, Glu, Leu, Lys, Val, and Ile residues were enriched in the water-soluble proteins, whereas in the transmembrane proteins, the Leu, Val, Ile, Phe, and Gly residues were enriched. To show the differences in amino acid content, the ratios of each amino acid of the water-soluble to the transmembrane proteins were calculated. In T. maritima, the 3 highest ratios were observed in Cys (3.29 = 0.92/0.28), Glu (2.16 = 10.04/
Table 1. Average amino acid compositions of 100 water-soluble and 100 transmembrane proteins with G + C content of genes.
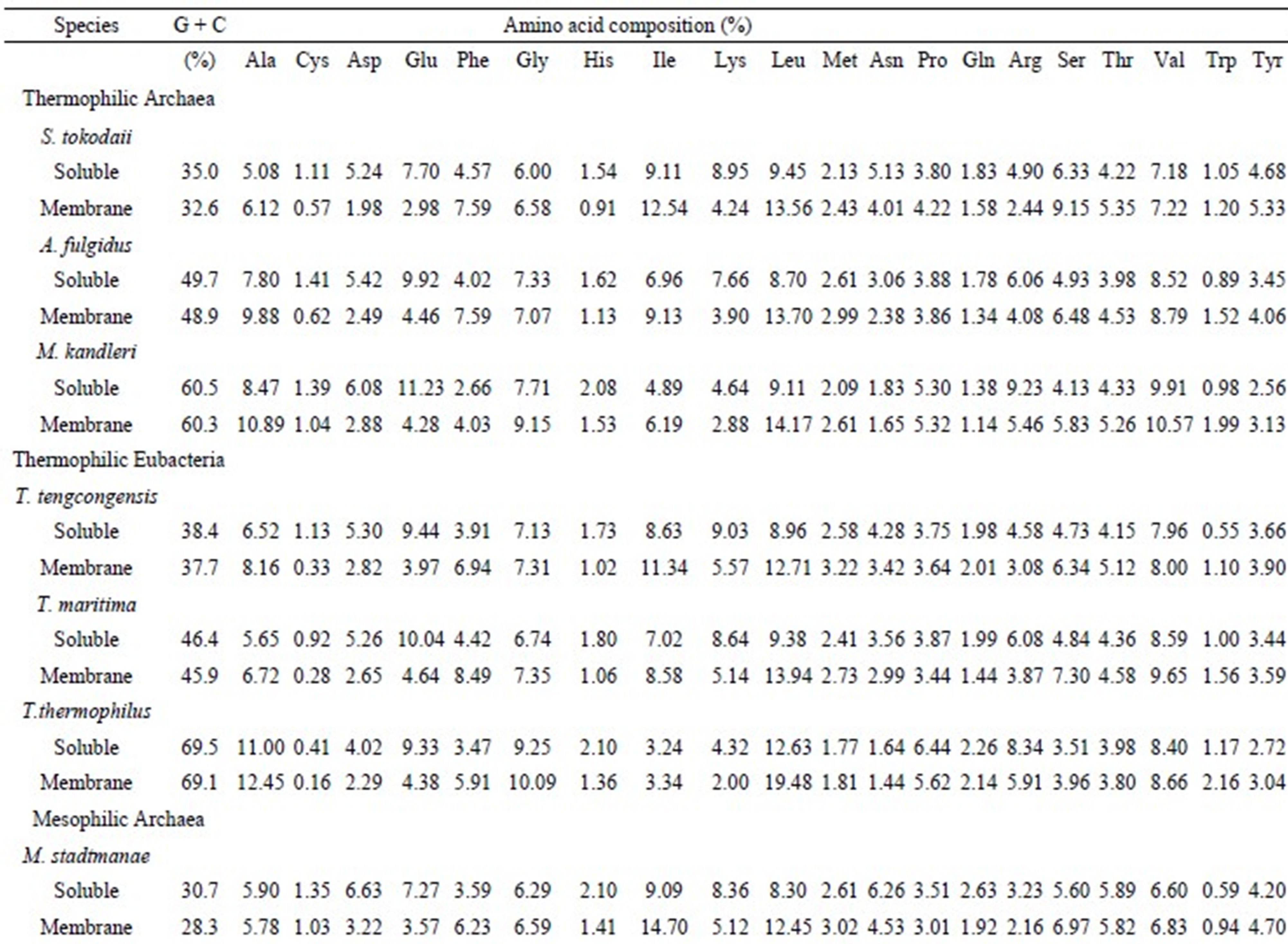
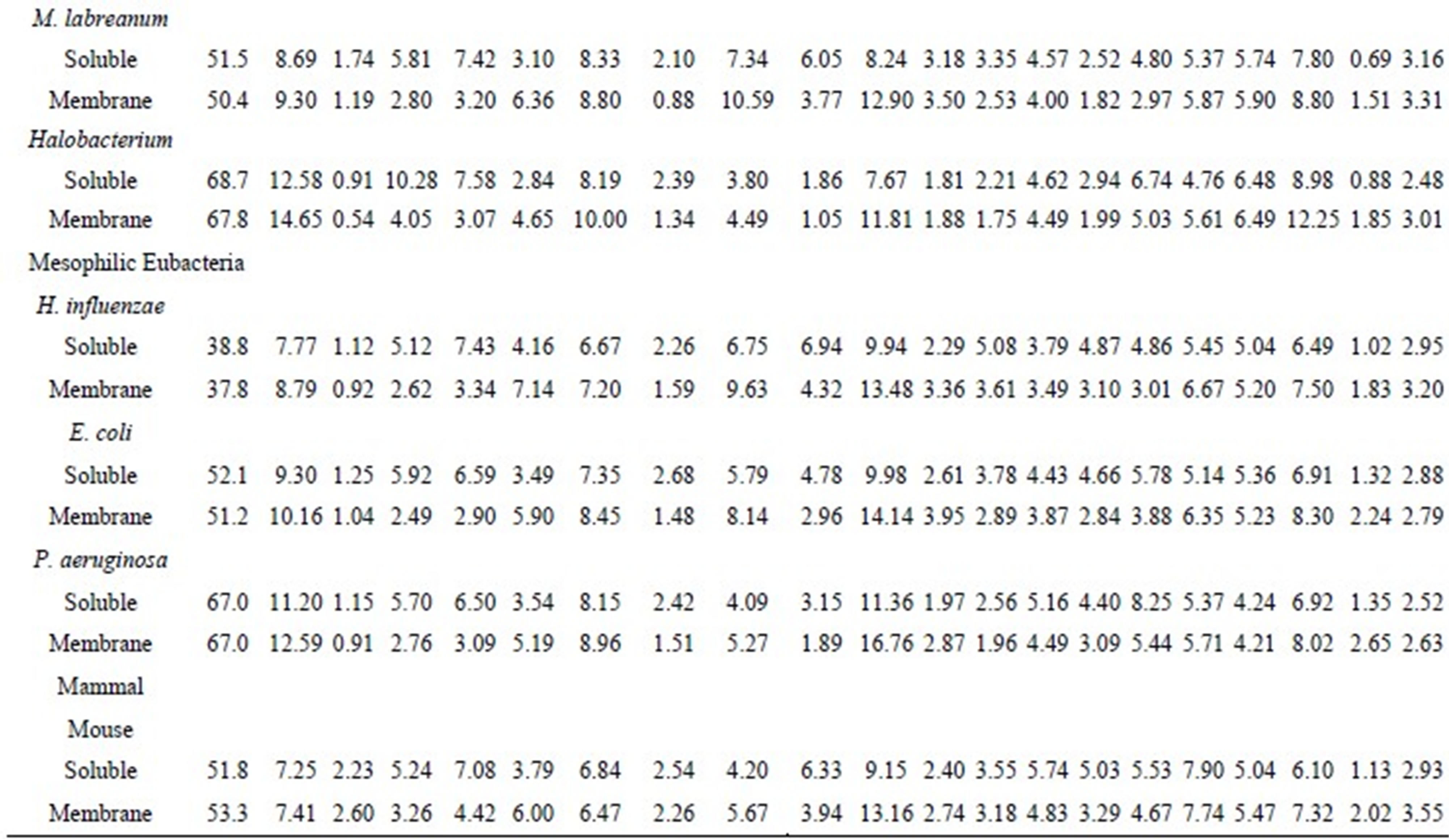
4.64), and Asp (1.98 = 5.26/2.65), while the 3 lowest ratios were Phe (0.52 = 4.42/8.49), Trp (0.64 = 1.00/1.56), and Ser (0.66 = 4.84/7.30). Frequently observed residues in the water-soluble and transmembrane proteins in the 12 species are listed in Table 2. In addition to Cys, the charged residues Glu, Asp, Lys, and His were frequently observed in water-soluble proteins. Comparatively, the hydrophobic Trp, Phe, Leu, and Met residues were frequently observed in transmembrane proteins. These results are not surprising as charged residues are suitable for water-soluble proteins, and hydrophobic residues are suitable for transmembrane proteins.
In addition, the frequency of some amino acid residues was dependent on the G + C content. For example, the frequency of Ala, Gly, and Pro residues, which are composed of G + C-rich codons, was increased in genes with high G + C content, whereas the frequency of Ile, Lys, and Asn residues, which are composed of A + T-rich codons, was decreased in genes with high G + C content. This tendency was observed in both water-soluble and transmembrane proteins, and it is consistent with previous findings [10-13,33]. The percentage of Lys and Ala residues in the water-soluble proteins plotted against G + C contents of genes demonstrated an almost linear relationship for proteins from both thermophiles and mesophiles (Figure 1). The Lys content was higher in the thermophilic proteins than in the mesophilic proteins, while the Ala content was the reverse. This is consistent with our previous findings that at higher temperature, DNA stability is enhanced by AA and decreased by GC [34]. The Lys content showed an almost linear relationship with the dinucleotide AA content, while Ala showed an almost linear relationship with the dinucleotide GC. The first and second nucleotides are AA in Lys codons, and GC in Ala codons. The genes encoding water-soluble proteins showed slightly higher G + C content than those encoding transmembrane proteins in all species, except P. aeruginosa (Table 1).
3.2. Favorable and Unfavorable Amino Acid Residues
The ratios of the observed to the expected amino acid compositions were calculated. Ratios of ≥1.3 were considered favorable and ≤0.7 were considered unfavorable. The favorable/unfavorable residues are listed in Table 3. In T. maritima, Glu, Phe, and Lys were favorable residues in both water-soluble and transmembrane proteins.
Table 2. List of amino acids frequently observed in the water-soluble and transmembrane proteins.
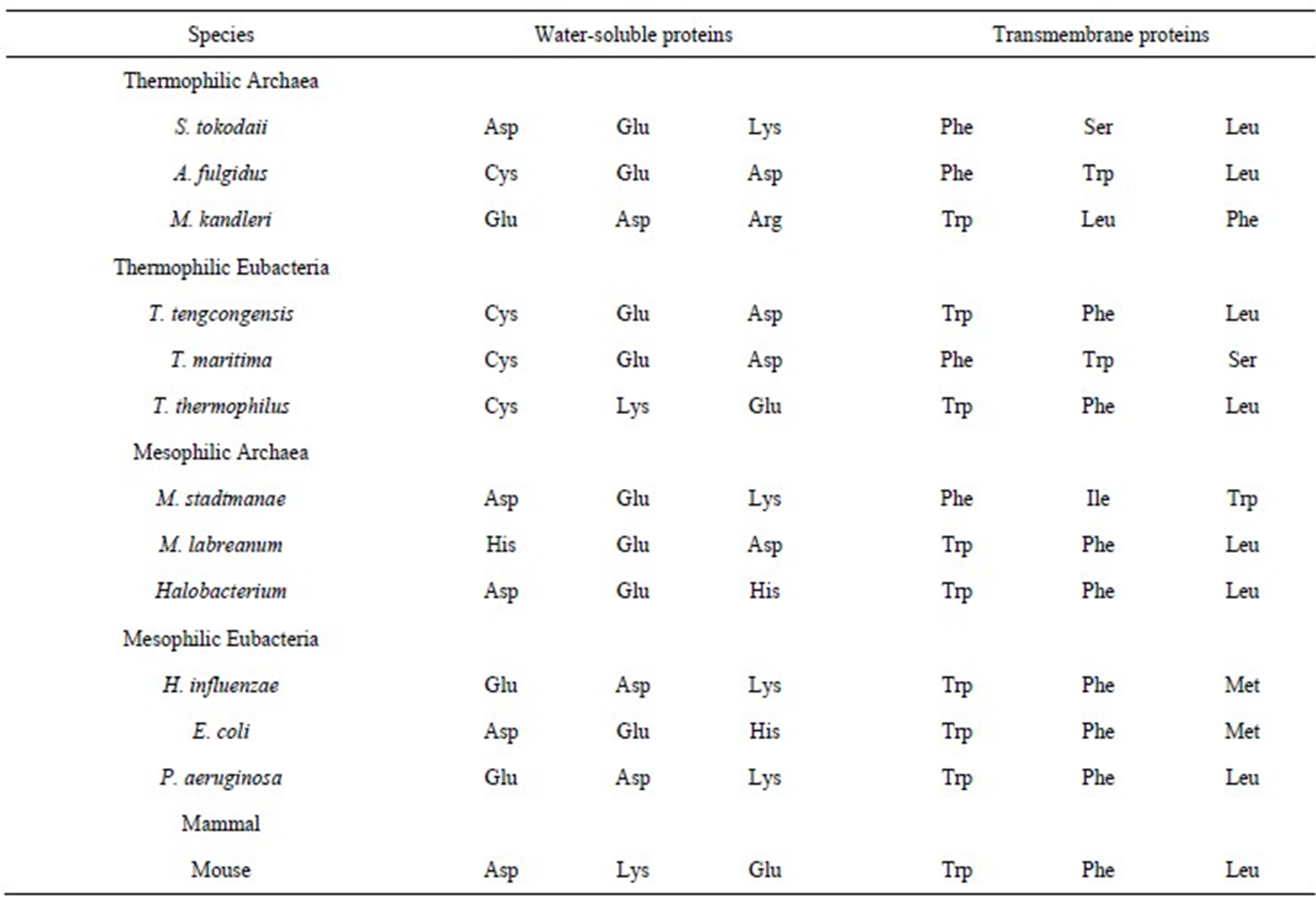
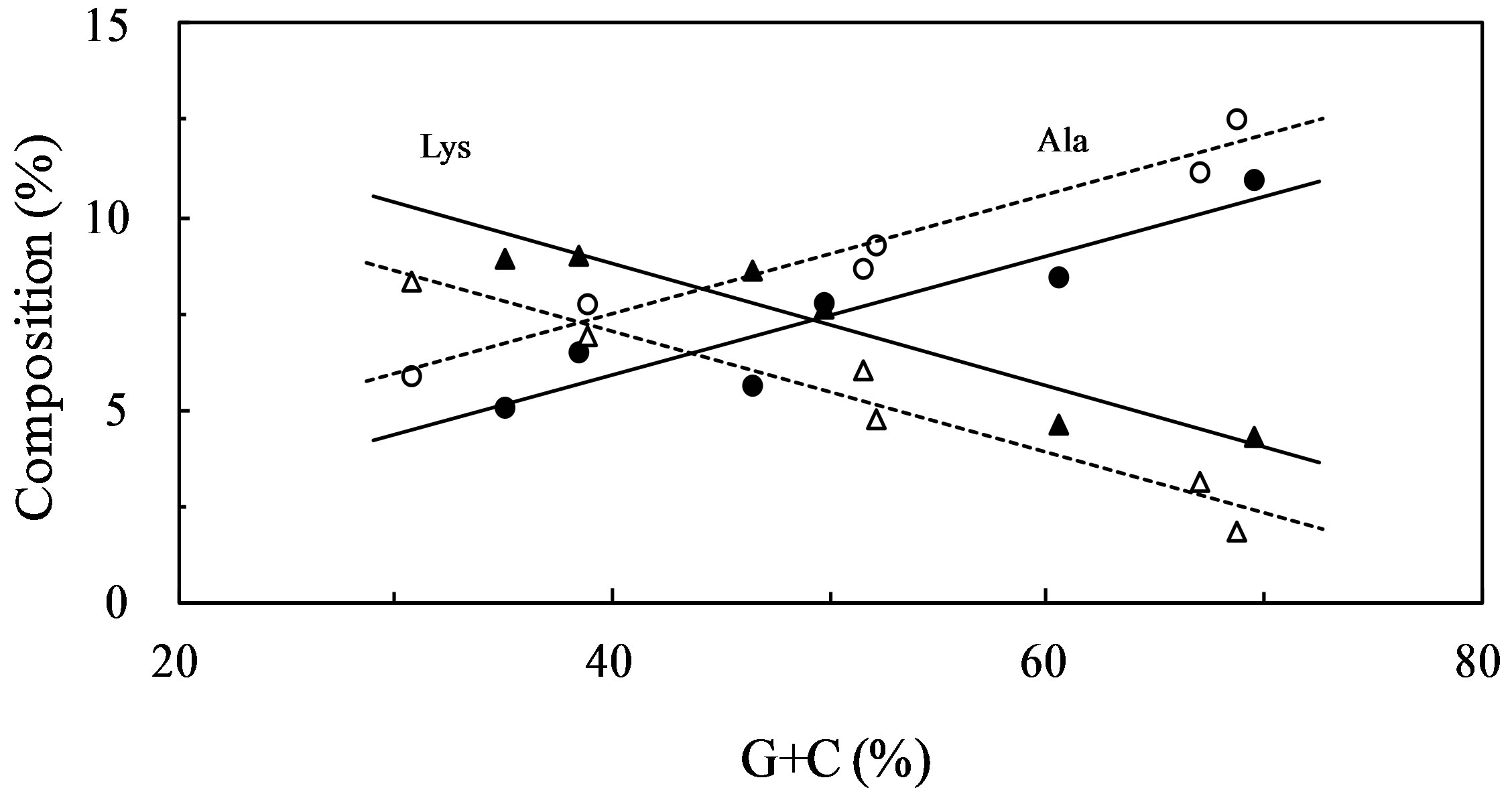
Figure 1. Linear correlation of Lys and Ala content with G + C content of the genes for water-soluble proteins. Solid and broken lines denote approximations for thermophilic and mesophilic proteins, respectively. Filled circles, Ala in thermophiles; open circles, Ala in mesophiles; filled triangles, Lys in thermophiles; open triangles, Lys in mesophiles.
Table 3. Favorable and unfavorable amino acids in the water-soluble and transmembrane proteins based on the ratios of observed/calculated composition.
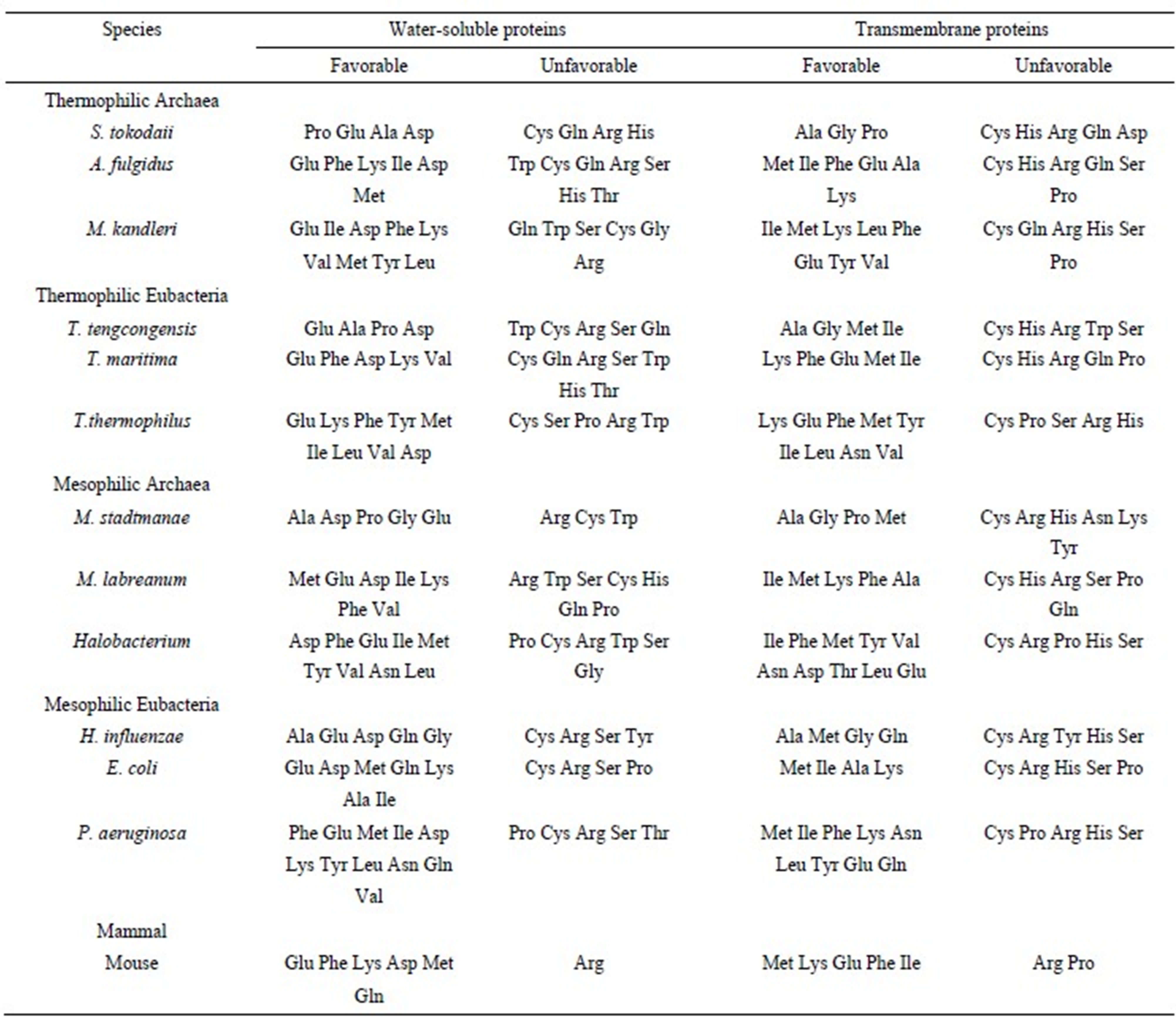
The percentage of Glu in water-soluble and transmembrane proteins was 10.04% and 4.64%, respectively, whereas that of Phe was 4.42% and 8.49%, respectively. Therefore, the amino acid compositions of Glu and Phe were different in the 2 protein groups, however, they were regarded as favorable in both the proteins. This is because the expected amino acid compositions were different for the 2 types of proteins due to the different nucleotide compositions of the 2 types of genes. In T. maritima, Cys, Gln, Arg, and His were estimated as unfavorable in both water-soluble and transmembrane proteins. Generally, the Glu, Asp, Lys, Ile, and Phe residues were favorable in the water-soluble proteins, and the Ile, Met, Phe, Ala, and Lys residues were favorable in the transmembrane proteins. A comparison of the amino acid compositions of the water-soluble and transmembrane proteins revealed that the Trp residue is abundant in the transmembrane proteins. However, Trp was not estimated as favorable. This result indicated that high proportions of an amino acid do not necessary dictate that it will be favorable. Glu and Asp were observed as favorable residues in all water-soluble proteins, whereas Cys and Arg were observed as unfavorable in both water-soluble and transmembrane proteins. No significant difference was observed with respect to favorable and unfavorable residues in thermophiles and mesophiles, with the exception of Gln. Consistent with the previous study [35], Gln was often observed as unfavorable in thermophiles.
The 3 highest ratios of observed/calculated composition were obtained for Asp in Halobacterium water-soluble proteins (3.54), Met in transmembrane proteins of P. aeruginosa (3.37), and Lys in transmembrane proteins of T. thermophilus (3.32); the former result was in agreement with previous study [19]. The 3 lowest ratios were calculated from the Cys content in the transmembrane proteins of the thermophilic Eubacteria T. thermophilus (0.04), T. maritima (0.07), and T. tengcongensis (0.09). This result is attributed to the very low observed Cys content in the transmembrane proteins of the thermophilic Eubacteria (Table 1).
The number of favorable residues in both water-soluble and transmembrane proteins increased with the G + C content; comparatively, the unfavorable residues did not. The Ala, Gly, and Pro residues, which have G + C-rich codons, were frequently observed as favorable in species with low G + C content. However, the Phe, Tyr, Lys, Ile, and Met residues, which have G + C-poor codons, were frequently observed as favorable in G + C-rich species. The positive correlation between the number of favorable residues and the G + C content may be due to the large number of residues that have G + C-poor codons, compared to those with G + C-rich codons. The Pro residue was observed as favorable in G + C-poor species, but was unfavorable in G + C-rich species. This result suggests that species maintain the Pro content in a certain range, increasing the supply when it is low and decreasing it when it is high.
Some species do not have aminoacyl tRNA synthetases for all 20 amino acids. For example, Halobacterium does not possess aminoacyl tRNA synthetases for Asn and Gln [27]. Generally, the Gln content reduces in the absence of aminoacyl tRNA synthetase for Gln, therefore, in these species, the Gln residue was regarded as unfavorable. Interestingly, the abundance of the Asn residue was not affected.
4. DISCUSSION
The amino acid sequences and compositions of watersoluble proteins differ from those of transmembrane proteins. The amino acid composition of a protein depends on the nucleotide sequence of the protein-coding gene. The average nucleotide composition of proteincoding genes from 3 animal mitochondria was A = 31%, C = 28%, G = 13%, and T = 28%. The proteins translated from the mitochondrial genes using the mitochondrial codon table [36] contain a significantly higher numbers of hydrophobic amino acid residues, therefore they are considered appropriate for transmembrane proteins. The observed amino acid composition correlated with the calculated amino acid content [37], indicating that designing proteins with specialized amino acid compositions is possible by a given specific nucleotide composition. In double-stranded DNA, the amount of adenine is equal to that of thymine, and the amount of guanine is equal to that of cytosine. This is known as Chargaff’s first parity rule [38,39]. This rule also applies to single-stranded DNA and is called Chargaff’s second parity rule [40,41]. This parity rule was confirmed by using over 3400 genomic sequences from Archaea, Eubacteria, eukaryotes, and viruses [42]. Species have to produce various kinds of proteins to survive under the constraints of Chargaff’s first and second parity rules for the DNA sequence. However, Chargaff’s second parity rule does not hold true for mitochondrial DNA [42].
The water-soluble and transmembrane proteins were obtained from the genes investigated in our previous study [4]. We examined the amino acid compositions of proteins from other species, and obtained similar trends corresponding to the G + C content. This result indicated that the characteristics of amino acid composition were maintained in proteins from various species. We selected species covering a wide range of G + C content, as it represents the mononucleotide composition. The amino acid composition is thought to be controlled by 2 factors, namely, the mononucleotide composition, and the deviation from expected values calculated using the mononucleotide values. Frequency of some amino acids is primarily dependent on G + C content as shown in Figure 1. Amino acids with high (or low) G + C content were frequently observed as favorable in species with low (or high) G + C content. These are the examples of the deviations from expectation to increase the supply when it is low.
Amino acids with a compositional ratio (observed/ calculated) of ≥1.3 were considered favorable and those with a compositional ratio of ≤0.7 were considered unfavorable. The ratios ranged from 0.04 to 3.54. Ratios of 1.1 and 0.9 were utilized to determine the favorable and unfavorable dinucleotides [4].
Both Arg and Cys residues were unfavorable in all the proteins from the Archaea and Eubacteria species. The depletion of the Arg residues in the amino acid sequences of mammals was identified more than 40 years ago [43]. In mammals, the cytosine of the dinucleotide CG is methylated to 5-methyl cytosine, which is more susceptible to deamination than cytosine that yields thymine. In addition, some of the T-G mismatches produced are poorly repaired, therefore, CG/CG tends to become TA/CA, which leads to a reduction in CG and an increase in TG and CA [44]. CG is a component of the Arg residue codon, CGN, and therefore, the repair-related errors lead to the depletion of Arg. To confirm this idea, we examined the amino acid sequences of both water-soluble and transmembrane proteins from mice.
The 100 water-soluble and 100 transmembrane proteins were selected according to the annotations of the GTOP database. The amino acid sequences having ≤30% sequence homology with other selected sequences were utilized. The nucleotide sequences corresponding to those protein sequences were retrieved from the NCBI web site, ftp://ftp.ncbi.nlm.nih.gov/genomes/M_musculus/RNA/rna.gbk.gz. The genes encoding water-soluble proteins were rich in AA, AG, and GA dinucleotides, whereas those encoding transmembrane proteins were rich in CT, TC, and TT. This trend was similar to that observed with the 12 species in our previous study [4]. The average amino acid compositions of the 100 water-soluble and 100 transmembrane proteins from mice are listed in Table 1, with the G + C content of the genes. The mouse genes encoding transmembrane proteins exhibited slightly higher G + C content than those encoding water-soluble proteins. The ratios of the amino acid composition of water-soluble proteins to those of transmembrane proteins were calculated. We observed a bias for Asp, Lys, and Glu residues in water-soluble proteins, and Trp, Phe, and Leu in transmembrane proteins (Table 2). This result was similar to that observed in the mesophilic species. Furthermore, Glu, Phe, Lys, and Met residues were favorable in both water-soluble and transmembrane proteins. However, only the Arg residue was deemed unfavorable in the mouse proteins (Table 3). The ratios of the observed to the expected dinucleotide compositions of CG, TG, and CA were 0.46, 1.39 and 1.23, respectively, for the genes encoding water-soluble proteins, and 0.48, 1.39, and 1.29, respectively, for the genes encoding transmembrane proteins. The ratios of CG (≤1) indicate lower amounts of CG in the genes, whereas the ratios of both TG and CA (≥1) indicate higher amounts of TG and CA. This result suggests that the CG/CG dinucleotides may now be TG/CA in the mouse genes. This trend was not seen in Archaea and Eubacteria, with the exception of M. stadtmanae. Therefore, the depletion of the Arg residue in Archaea and Eubacteria might be due to different reasons compared to those responsible in mammals.
5. ACKNOWLEDGEMENTS
This study was supported in part by Japan Society for the Promotion of Science, Research (C) No. 21500681 to K.Kitamura.
REFERENCES
- Kyte, J. and Doolittle, R.F. (1982) A simple method for displaying the hydropathic character of a protein. Journal of Molecular Biology, 157, 105-132. doi:10.1016/0022-2836(82)90515-0
- Klein, P., Kanehisa, M. and DeLisi, C. (1985) The detection and classification of membrane-spanning proteins. Biochimica et Biophysica Acta-Biomembranes, 815, 468- 476. doi:10.1016/0005-2736(85)90375-X
- Hirokawa, T., Boon-Chieng, S. and Mitaku, S. (1998) SOSUI: Classification and secondary structure prediction system for membrane proteins. Bioinformatics, 14, 378- 379. doi:10.1093/bioinformatics/14.4.378
- Nakashima, H. and Kuroda, Y. (2011) Differences in dinucleotide frequencies of thermophilic genes encoding water soluble and membrane proteins. Journal of Zhejiang University-Science B (Biomedicine & Biotechnology), 12, 419-427.
- Muto, A. and Osawa, S. (1987) The guanine and cytosine content of genomic DNA and bacterial evolution. Proceedings of the National Academy of Sciences of the United States of America, 84, 166-169. doi:10.1073/pnas.84.1.166
- Lawrence, J.G. and Ochman, H. (1997) Amelioration of bacterial genomes: rates of change and exchange. Journal of Molecular Evolution, 44, 383-397. doi:10.1007/PL00006158
- Karlin, S. and Burge, C. (1995) Dinucleotide relative abundance extremes: A genomic signature. Trends in Genetics, 11, 283-290. doi:10.1016/S0168-9525(00)89076-9
- Karlin, S., Mrázek, J. and Campbell, A.M. (1997) Compositional biases of bacterial genomes and evolutionary implications. Journal of Bacteriology, 179, 3899-3913.
- Nakashima, H., Ota, M., Nishikawa, K. and Ooi, T. (1998) Gene from nine genomes are separated into their organisms in the dinucleotide composition space. DNA Research, 5, 251- 259. doi:10.1093/dnares/5.5.251
- Singer, G.A.C. and Hickey, D.A. (2000) Nucleotide bias causes a genomewide bias in the amino acid composition of proteins. Molecular Biology and Evolution, 17, 1581- 1588. doi:10.1093/oxfordjournals.molbev.a026257
- Bharanidharan, D., Bhargavi, G.R., Uthanumallian, K. and Gautham, N. (2004) Correlations between nucleotide frequencies and amino acid composition in 115 bacterial species. Biochemical and Biophysical Research Communications, 315, 1097-1103. doi:10.1016/j.bbrc.2004.01.129
- Hu, J., Zhao, X., Zhang, Z. and Yu, J. (2007) Compositional dynamics of guanine and cytochine content in prokaryotic genomes. Research in Microbiology, 158, 363- 370. doi:10.1016/j.resmic.2007.02.007
- Lobry, J.R. (1997) Influence of genomic G+C content on average amino-acid composition of proteins from 59 bacterial species. Gene, 205, 309-316. doi:10.1016/S0378-1119(97)00403-4
- Kumar, S., Tsai, C.J. and Nussinov, R. (2000) Factors enhancing protein thermostability. Protein Engineering, 13, 179-191. doi:10.1093/protein/13.3.179
- Kreil, D.P. and Ouzounis, C.A. (2001) Identification of thermophilic species by the amino acid compositions deduced from their genomes. Nucleic Acids Research, 29, 1608-1615. doi:10.1093/nar/29.7.1608
- Farias, S.T. and Bonato, M.C.M. (2003) Preferred amino acids and thermostability. Genetics and Molecular Research, 2, 383-393.
- Yokota, K., Satou, K. and Ohki, S. (2006) Comparative analysis of protein thermostability: Differences in amino acid content and substitution at the surfaces and in the core regions of thermophilic and mesophilic proteins. Science and Technology of Advanced Materials, 7, 255- 262. doi:10.1016/j.stam.2006.03.003
- Zhou, X.-X., Wang, Y.-B., Pan, Y.-J. and Li, W.-F. (2008) Differences in amino acids composition and coupling patterns between mesophilic and thermophilic proteins. Amino Acids, 34, 25-33. doi:10.1007/s00726-007-0589-x
- Fukuchi, S., Yoshimune, K., Wakayama, M., Moriguchi, M. and Nishikawa, K. (2003) Unique amino acid composition of proteins in halophilic bacteria. Journal of Molecular Biology, 327, 347-357. doi:10.1016/S0022-2836(03)00150-5
- Kawarabayasi, Y., Hino, Y., Horikawa, H., Jin-no, K., Takahashi, M., Sekine, M., Baba, S., Ankai, A., Kosugi, H., Hosoyama, A., Fukui, S., Nagai, Y., Nishijima, K., Otsuka, R., Nakazawa, H., Takamiya, M., Kato, Y., Yoshizawa, T., Tanaka, T., Kudoh, Y., Yamazaki, J., Kushida, N., Oguchi, A., Aoki, K., Masuda, S., Yanagii, M., Nishimura, M., Yamagishi, A., Oshima, T. and Kikuchi, H. (2001) Complete genome sequence of an aerobic thermoacidophilic crenarchaeon, Sulfolobus tokodaii strain 7. DNA Research, 8, 123-140. doi:10.1093/dnares/8.4.123
- Klenk, H.-P., Clayton, R.A., Tomb, J.-F., White, O., Nelson, K.E., Ketchum, K.A., Dodson, R.J., Gwinn, M., Hickey, E.K., Peterson, J.D., Richardson, D.L., Kerlavage, A.R., Graham, D.E., Kyrpides, N.C., Fleischmann, R.D., Quackenbush, J., Lee, N.H., Sutton, G.G., Gill, S., Kirkness, E.F., Dougherty, B.A., McKenney, K., Adams, M.D., Loftus, B., Peterson, S., Reich, C.I., McNeil, L.K., Badger, J.H., Glodek, A., Zhou, L., Overbeek, R., Gocayne, J.D., Weidman, J.F., McDonald, L., Utterback, T., Cotton, M.D., Spriggs, T., Artiach, P., Kaine, B.P., Sykes, S.M., Sadow, P.W., D’Andrea, K.P., Bowman, C., Fujii, C., Garland, S.A., Mason, T.M., Olsen, G.J., Fraser, C.M., Smith, H.O., Woese, C.R. and Venter, J.C. (1997) The complete genome sequence of the hyperthermophilic, sulphate-reducing archaeon Archaeoglobus fulgidus. Nature, 390, 364-370. doi:10.1038/37052
- Slesarev, A.I., Mezhevaya, K.V., Makarova, K.S., Polushin, N.N., Shcherbinina, O.V., Shakhova, V.V., Belova, G.I., Aravind, L., Natale, D.A., Rogozin, I.B., Tatusov, R.L., Wolf, Y.I., Stetter, K.O., Malykh, A.G., Koonin, E.V. and Kozyavkin, S.A. (2002) The complete genome of hyperthermophile Methanopyrus kandleri AV19 and monophyly of archaeal methanogens. Proceedings of the National Academy of Sciences of the United States of America, 99, 4644-4649. doi:10.1073/pnas.032671499
- Bao, Q., Tian, Y., Li, W., Xu, Z., Xuan, Z., Hu, S., Dong, W., Yang, J., Chen, Y., Xue, Y., Xu, Y., Lai, X., Huang, L. Dong, X., Ma, Y., Ling, L., Tan, H., Chen, R., Wang, J., Yu, J. and Yang, H. (2002) A complete sequence of the T. tengcongensis genome. Genome Research, 12, 689- 700. doi:10.1101/gr.219302
- Nelson, K.E., Clayton, R.A., Gill, S.R., Gwinn, M.L., Dodson, R.J., Haft, D.H., Hickey, E.K., Peterson, J.D., Nelson, W.C., Ketchum, K.A., McDonald, L., Utterback, T.R., Malek, J.A., Linher, K.D., Garrett, M.M., Stewart, A.M., Cotton, M.D., Pratt, M.S., Phillips, C.A., Richardson, D., Heidelberg, J., Sutton, G.G., Fleischmann, R.D., Eisen, J.A., White, O., Salzberg, S.L., Smith, H.O., Venter, J.C. and Fraser, C.M. (1999) Evidence for lateral gene transfer between Archaea and Bacteria from genome sequence of Thermotoga maritima. Nature, 399, 323-329. doi:10.1038/20601
- Fricke, W.F., Seedorf, H., Henne, A., Krüer, M., Liesegang, H., Hedderich, R., Gottschalk, G. and Thauer, R.K. (2006) The genome sequence of Methanosphaera stadtmanae reveals why this human intestinal archaeon is restricted to methanol and H2 for methane formation and ATP synthesis. Journal of Bacteriology, 188, 642-658. doi:10.1128/JB.188.2.642-658.2006
- Anderson, I., Ulrich, L.E., Lupa, B., Susanti, D., Porat, I., Hooper, S.D., Lykidis, A., Sieprawska-Lupa, M., Dharmarajan, L., Goltsman, E., Lapidus, A., Saunders, E., Han, C., Land, M., Lucas, S., Mukhopadhyay, B., Whitman, W.B., Woese, C., Bristow, J. and Kyrpides, N. (2009) Genomic characterization of methanomicrobiales reveals three classes of methanogens. PLoS One, 4, 1-9. doi:10.1371/journal.pone.0005797
- Ng, W.V., Kennedy, S.P., Mahairas, G.G., Berquist, B., Pan, M., Shukla, H.D., Lasky, S.R., Baliga, N.S., Thorsson, V., Sbrogna, J., Swartzell, S., Weir, D., Hall, J., Dahl, T.A., Welti, R., Goo, Y.A., Leithauser, B., Keller, K., Cruz, R., Danson, M.J., Hough, D.W., Maddocks, D.G., Jablonski, P.E., Krebs, M.P., Angevine, C.M., Dale, H., Isenbarger, T.A., Peck, R.F., Pohlshroder, M., Spudich, J.L., Jung, K-H., Alam, M., Freitas, T., Hou, S., Daniels, C.J., Dennis, P.P., Omer, A.D., Ebhardt, H., Lowe, T.M., Liang, P., Riley, M., Hood, L. and DasSarma S. (2000) Genome sequence of Halobacterium species NRC-1. Proceedings of the National Academy of Sciences of the United States of America, 97, 12176-12181. doi:10.1073/pnas.190337797
- Fleischmann, R.D., Adams, M.D., White, O., Clayton, R.A., Kirkness, E.F., Kerlavage, A.R., Bult, C.J., Tomb, J.-F., Dougherty, B.A., Merrick, J.M., Mckenney, K., Sutton, G., FitzHugh, W., Fields, C., Gocayne, J.D., Scott, J., Shirley, R., Liu, L.-I., Glodek, A., Kelley, J.M., Weidman, J.F., Philips, C.A., Spriggs, T., Hedbolm, E., Cotton, M.D., Utterback, T.R., Hanna, M.C., Nguyen, D.T., Saudek, D.M., Brandon, R.C., Fine, L.D., Fritchman, J.L., Fuhrmann, J.L., Geoghagen, N.S.M., Gnehm, C.L., McDonald, L.A., Small, K.V., Fraser, C.M., Smith, H.O. and Venter, J.C. (1995) Whole-genome random sequencing and assembly of Haemophilus influenzae rd. Science, 269, 496-512. doi:10.1126/science.7542800
- Blattner, F.R., Plunkett, G.III., Bloch, C.A., Perna, N.T., Burland, V., Riley, M., Collado-Vides, J., Glasner, J.D., Rode, C.K., Mayhew, G.F., Gregor, J., Davis, N.W., Kirkpatrick, H.A., Goeden, M.A., Rose, D.J., Mau, B. and Shao, Y. (1997) The complete genome sequence of Escherichia coli K-12. Science, 277, 1453-1462. doi:10.1126/science.277.5331.1453
- Stover, C.K., Pham, X.Q., Erwin, A.L., Mizoguchi, S.D., Warrener, P., Hickey, M.J., Brinkman, F.S.L., Hufnagle, W.O., Kowalik, D.J., Lagrou, M., Garber, R.L., Goltry, L., Tolentino, E., Westbrock-Wadman, S., Yuan, Y., Brody, L.L., Coulter, S.N., Folger, K.R., Kas, A., Larbig, K., Lim, R., Smith, K., Spencer, D., Wong, G.K.-S., Wu, Z., Paulsen, I.T., Reizer, J., Saier, M.H., Hancock, R.E.W., Lory, S. and Olson, M.V. (2000) Complete genome sequence of Pseudomonas aeruginosa PA01, an opportunistic pathogen. Nature, 406, 959-964. doi:10.1038/35023079
- Kawabata, T., Fukuchi, S., Homma, K., Ota, M., Araki, J., Ito, T., Ichiyoshi, N. and Nishikawa, K. (2002) GTOP: A database of protein structures predicted from genome sequences. Nucleic Acids Research, 30, 294-298. doi:10.1093/nar/30.1.294
- Altschul, S.F., Gish, W., Miller, W., Myers, E.W. and Lipman, D.J. (1990) Basic local alignment search tool. Journal of Molecular Biology, 215, 403-410.
- Wilquet, V. and Van de Casteele, M. (1999) The role of the codon first letter in the relationship between genomic GC content and protein amino acid composition. Research in Microbiology, 150, 21-32. doi:10.1016/S0923-2508(99)80043-6
- Nakashima, H., Fukuchi, S. and Nishikawa, K. (2003) Compositional changes in RNA, DNA and proteins for bacterial adaptation to higher and lower temperatures. The Journal of Biochemistry, 133, 507-513. doi:10.1093/jb/mvg067
- Jaenicke, R. and Bӧhm, G. (1998) The stability of proteins in extreme environments. Current Opinion in Structural Biology, 8, 738-748. doi:10.1016/S0959-440X(98)80094-8
- Barrel, B.G., Anderson, S., Bankier, A.T., de Bruijn, M.H.L., Chen, E., Coulson, A.R., Drouin, J., Eperon, I.C., Nerlich, D.P., Roe, B.A., Sanger, F., Schreier, P.H., Smith, A.J.H., Staden, R. and Young I.G. (1980) Different pattern of codon recognition by mammalian mitochondrial tRNAs. Proceedings of the National Academy of Sciences of the United States of America, 77, 3164- 3166. doi:10.1073/pnas.77.6.3164
- Nakashima, H., Nishikawa, K. and Ooi, T. (1990) Distinct character in hydrophobicity of amino acid compositions of mitochondrial proteins. Proteins, 8, 173-178. doi:10.1002/prot.340080207
- Chargaff, E., Lipshitz, R., Green, C. and Hodes, M.E. (1951) The composition of the desoxyribonucleic acid of salmon sperm. The Journal of Biological Chemistry, 192, 223-230.
- Chargaff, E., Lipshitz, R. and Green, C. (1952) Composition of the desoxypentose nucleic acids of four genera of sea-urchin. The Journal of Biological Chemistry, 195, 155-160.
- Karkas, J.D., Runder, R. and Chargaff, E. (1968) Separation of B. subtilis DNA into complementary strands, II. Template functions and composition as determined by transcription with RNA polymerase. Proceedings of the National Academy of Sciences of the United States of America, 60, 915-920. doi:10.1073/pnas.60.3.915
- Runder, R., Karkas, J.D. and Chargaff, E. (1968) Separation of B. subtilis DNA into complementary strands, III. Direct analysis. Proceedings of the National Academy of Sciences of the United States of America, 60, 921-922. doi:10.1073/pnas.60.3.921
- Mitchell, D. and Bridge, R. (2006) A test of Chargaff’s second rule. Biochemical and Biophysical Research Communications, 340, 90-94. doi:10.1016/j.bbrc.2005.11.160
- King, J.L. and Jukes, T.H. (1969) Non-Darwinian evolution. Science, 164, 788-798. doi:10.1126/science.164.3881.788
- Lutsenko, E. and Bhagwat, A.S. (1999) Principal causes of hot spots for cytosine to thymine mutations at sites of cytosine methylation in growing cells: A model, its experimental support and implications. Mutation Research, 437, 11-20. doi:10.1016/S1383-5742(99)00065-4