Journal of Cancer Therapy
Vol.3 No.4(2012), Article ID:21589,11 pages DOI:10.4236/jct.2012.34035
Preclinical Efficacy of Nimotuzumab, an Anti-EGFR Monoclonal Antibody as a Single Agent Therapy in Human GBM U87MG Xenografts
![]()
1Center of Molecular Immunology, Havana, Cuba; 2Max Delbrück Center for Molecular Medicine, Berlin, Germany.
Email: *arlhee@cim.sld.cu
Received June 2nd, 2012; revised July 6th, 2012; accepted July 18th, 2012
Keywords: CD133; Epidermal Growth Factor Receptor; Glioblastoma Multiforme; Nimotuzumab; Radiation; Temozolomide
ABSTRACT
Background: The poor prognosis of patients with high-grade glioma multiform (GBM) has led investigators to the search of new therapeutic strategies. Current treatment includes surgery when possible, radiotherapy and chemotherapy. Molecular-targeted therapies are in the process of clinical testing, and promising agents include monoclonal antibodies. Our study examined the antitumor activity of three different single therapies in nude mice bearing both subcutaneous and orthotopic brain xenografts of the U87MG human GBM cell line. Methods: Cell culture, Histology, Immunohistochemistry, Animal experiments, Statistical analysis. Results: Different groups of treatment included nimotuzumab, a humanized monoclonal antibody that inhibits the EGFR tyrosine kinase activity, or total body irradiation, or the chemotherapeutic agent temozolomide (TMZ). For the control group animals received saline solution instead of the antibody. For the subcutaneous model, only nimotuzumab or TMZ produced a significant delay in tumor growth. In the intracranial model, unlike TMZ, the systemic administration of the antibody did not reduce the tumor growth, despite both therapies inhibited the formation of microsatellites in the brain of mice. The antitumor activity of nimotuzumab was accompanied by a decrease in the microvessel density and the proliferative activity of tumor cells. TMZ only inhibited the tumor cell proliferation but not the formation of new tumor-associated microvessels in xenografts. For radiation therapy, neither antiproliferative nor antiangiogenic activity was found, in accordance with the lack of antitumor activity. Only nimotuzumab reduced the frequency of chemo and radioresistant CD133+ population. Conclusion: Our results illustrate the potential efficacy of nimotuzumab as a single agent against an EGFR-amplified human GBM, a tumor resistant to the therapy with all well-known forms of treatment.
1. Introduction
Human glioblastomas are the most lethal primary brain tumor with a median survival of 14.6 months despite aggressive treatment using surgery, radiation, and chemotherapy [1,2]. The scarce efficacy of current available therapies is primarily attributable to the de novo or acquired resistance to radiation and chemotherapeutic agents [3]. Among the currently available therapies ionizing radiation has been probably the most effective nonsurgical treatment modality. Nevertheless, virtually all patients with GBM have an inadequate response to treatment, with frequent relapses or progresses after radiation. Recurrent tumors are highly aggressive and resistant to other forms of therapies and invariably lead to patient death in a short period of time [4]. Adjuvant chemotherapy with singleagents or combination therapy is also far from satisfactory. Although alkylating agents such as TMZ have activity as single-agents or in combination chemotherapy, its use has been associated with a high level toxicity and only a modest improvement in overall survival rate [5,6]. In general, these agents are severely cytotoxic or poorly tolerated, and resistance develops rapidly, which limits the minimal benefits offered by treatment with these drugs [5]. As a result, there is a need for new agents that are effective and can be used in combination with conventional therapies to overcome resistance, improve local control of the disease and overall survival of patients with GBM [7].
Most of high-grade gliomas overexpress the Epidermal Growth Factor Receptor (EGFR) and this overexpression is usually associated with a more aggressive phenotype and a worse clinical outcome. This has led to developing new molecular-targeted therapies based on the inhibition of the EGFR. Among EGFR inhibitors, the humanized anti-EGFR monoclonal antibody nimotuzumab (TheraCIM, h-R3) has undergone an extensive evaluation in GBM [8,9]. Preliminary clinical studies conducted with nimotuzumab demonstrated its efficacy and low toxicity profile in the treatment of patients with this malignancy [8]. The good tolerability of this drug has particular clinical value for the treatment of pediatric glioma, for which chemotherapy is often the primary modality [10]. Moreover, its favorable safety profile allows it to be coadministered with various agents, and most important give the possibility to be used under long-term schemes without a dose-limiting toxicity [8,10]. Based on this data, investigators have initiated a phase III trial to confirm the activity of nimotuzumab concomitant with RT in this type of tumor.
Previous studies conducted by our group have demonstrated the ability of nimotuzumab to potentiate the activity of radiation in the human GBM cell line U87MG [10]. However, its potential efficacy as a single agent therapy has not been fully addressed by preclinical studies. Such study is mandatory to provide preclinical evidence for the benefit of nimotuzumab in GMB, in addition to give further insights about the mechanisms of action of this drug for the design of therapeutic combinations with standard cytotoxic agents. In addition, such study might illustrate the potential therapeutic efficacy of nimotuzumab as a successful alternative of therapy in GBM patients not amenable to other forms of treatment. We therefore investigated the efficacy of a nimotuzumab therapy in comparison to other single-agents treatments such as radiation and chemotherapy with TMZ, evaluating its growth-inhibitory effects and potential mechanisms of antitumor activity in a human GBM cell line xenografted in NMRI nude mice.
2. Materials and Methods
Cell Culture. U87MG (HTB-14, ATCC) is a human GBM cell line. Cells were grown in a 1 + 1 mixture of Earle’s MEM and Basal medium (Sigma) containing 2 mM L-glutamine and 10% fetal bovine serum, and were maintained under a humidified atmosphere of 5% CO2 at 37˚C.
Chemicals and Antibodies. Temozolomide was purchased from Schering-Plough (stock solution 150 mmol/L in DMSO). The humanized anti-EGFR mAb nimotuzumab was generated at the Center of Molecular Immunology [11,12]. All the primary and secondary antibodies were purchased from commercial sources as listed: rabbit polyclonal EGFR antibody to total EGFR (Santa Cruz Biotechnology), mouse monoclonal antibody MIB-1 to Ki-67 (DakoCytomation), rat monoclonal anti CD31/ PECAM-1 antibody (BD Pharmingen), mouse monoclonal antibody to CD133/1 (AC133) (Miltenyi Biotec). The secondary antibodies used were: HRP-conjugated anti-rat IgG1 (Southern Biotech), HRP-conjugated antirabbit IgG (DakoCytomation), HRP-conjugated antimouse IgG (DakoCytomation).
Histology and Immunohistochemistry. All the specimens were fixed in 4% paraformaldehyde, embedded in Tissue-Tek OCT (optimal cutting temperature) compound and frozen at –20˚C until analysis. For histologic examination, tissue slides (5 µm in thickness) were stained with hematoxylin and eosin. For immunohistochemistry, tissue sections (thickness of 8 µm) were immunostained according to standard protocols. To analyze the proliferative activity of the tumor cells, sections were stained with the MIB-1 antibody (1:50) against the Ki-67 as described previously [11]. The percentage of MIB-1-positive nuclei was determined by counting immunoreactive tumor cell nuclei in at least 5 high-power fields in the most actively proliferating tumor area. To score a tumor cell as positive for Ki-67, a nuclear staining was required. To detect microvessels, sections were stained with an antibody against CD31/PECAM-1 (1:100). Vessel density was determined by counting the number of stained vessels in three to five high-power fields (0.031 mm2) in the most densely vascularized areas. To detect CD133 positive cells, tumor sections were stained with the anti-human CD133/1 (AC133) (1:10) as described [11]. To determine EGFR expression tumor sections were stained as described [11]. Negative controls consisted of duplicate sections of the same specimens in which the primary antibody had been excluded and replaced with PBS or negative control immunoglobulin. Sections were visualized with 3,3’-diaminobenzidine as a chromogen and counterstained with Mayer’s hematoxylin. Representative tumor sections were identified on a light microscope (Zeiss, Axioskop 40) with an ocular magnification of ×40 evaluating 4 to 5 tumors from each group for the corresponding analysis. To score a tumor cell as positive a complete membrane staining was required for EGFR and CD133.
Animal Experiments. Female athymic mice (8 - 10 weeks old, nu/nu) were obtained from Charles River. The mice were housed and maintained under aseptic conditions in facilities approved by the German Association for Accreditation of Laboratory Animal Care and in accordance with current regulations and standards of the German Animal Protection Law, and their use was approved by the local responsible authorities. Animals met the requirements of the UKCCCR guidelines [13]. To produce xenografts, tumor cells were harvested from subconfluent cultures by treatment with 0.25% trypsin and 0.05% EDTA. Only single-cell suspensions with >90% viability were used for injections. Animals were inoculated with both 107 U87MG tumor cells s.c. into left flank and 2 × 104 cells intracranially into the right hemisphere of mouse brains with the help of a stereotactic device. Tumor volume from flank’s were determined from direct measurement with calipers and calculated according to the formula: 0.5 × (large diameter) × (small diameter)2. Relative tumor volumes (RTV) were calculated by referring the median volumes of each day to the first measurement (set to 1) as described [14]. Treatments were initiated three days after tumor cell injection. Treatment groups consisted of control, anti-EGFR monoclonal antibody nimotuzumab, total body irradiation, and chemotherapy with TMZ, with each group containing eight mice, except the control group containing ten mice. The antibody was administered intraperitoneal three times per week with 1 mg per mouse (50 mg/kg), mice in the control; radiation and TMZ groups were injected with saline solution. For radiation group, animals were exposed to a total dose of 4.0 Gy of total body radiation fractioned in 1.0 Gy weekly. For chemotherapy group, animals received five doses of TMZ 200 µg (10 mg/kg) in consecutive days, starting 3 days after tumor inoculation. All animals were killed by day 31 when tumor weight from the control group exceeded the ten percent of total animal weight.
The size of intracranial (i.c.) tumors was determined as described previously [11]. At the area of largest dimensions tumor diameters and perimeters were determined by computer-assisted image with the help of a microscope (Zeiss Axioskop 40). Subcutaneous tumors were snapping frozen and stored at –80˚C for additional analyses.
Statistical Analysis. Statistical analysis was performed using the GraphPAD Prism software for Windows, version 4.0 (GraphPAD). The significance of differences between groups was compared using a Kruskal-Wallis test. The significance of differences in groups was compared using Turkey-Kramer’s Multiple Comparison Test. Differences were considered significant if p < 0.05.
3. Results
We first explored the antitumor activity of different single-agent therapies (e.g. total body irradiation, chemotherapy with TMZ or molecular-targeted therapy with nimotuzumab) on the growth of U87MG tumors. For that purposes U87MG cells were xenografted both in the flanks and in the brains of NMRI nude mice and antitumor activities achieved with each therapy were assessed. All animals injected with the U87MG cells developed tumors. Figure 1 shows the relative tumor volumes (RTV) of treated groups at different time points. As shown, only the treatment with TMZ or nimotuzumab caused a substantial growth delay and subsequent inhibition of the growth rate of U87MG xenografts with a maximum effect by day 28, i.e., media RTV for TMZ (1.7) and nimotuzumab (1.4) versus control (3.1). In contrast, the exposure to ionizing radiation did not affect tumor growth profiles in treated animals compared with controls (RTV of 2.4 by day 28). These results suggest that single agents nimotuzumab and TMZ had higher antitumor activity than ionizing radiation in U87MG tumors in vivo under the dose and schedule assessed.
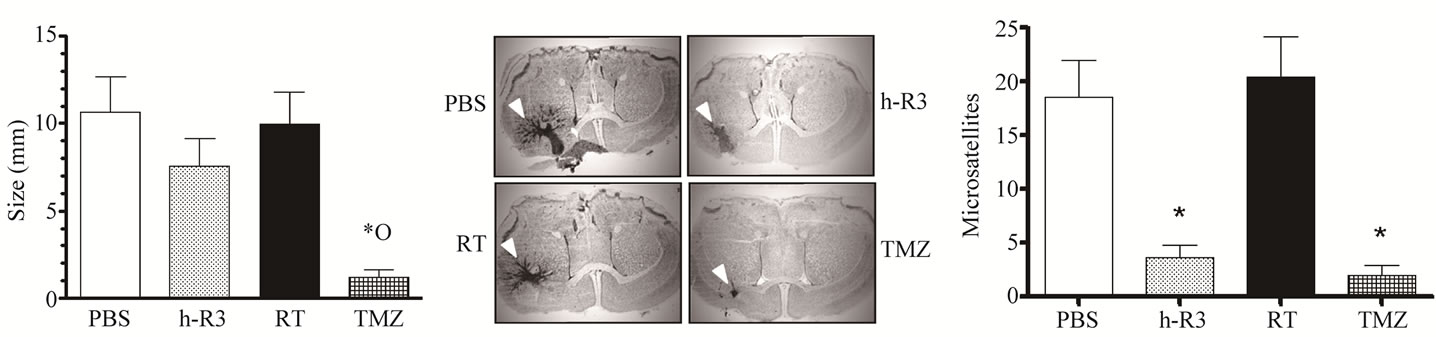
We next examined the antitumor activity of each therapy in U87MG tumors implanted in the brain of mice. In the orthotopic model, radiation failed to show a statistical reduction in brain tumor size (p > 0.05), corroborating its lack of efficacy previously observed in subcutaneous (s.c.) tumors (Figure 2(a)). Surprisingly, in brain tumors nimotuzumab did not reproduce the efficacy observed in s.c. tumors, and despite slight reductions in tumor size, a non significant activity was found; e.g. tumor perimeter (mean ± sem) for nimotuzumab (7.6 mm ± 1.6) versus radiotherapy (10.0 mm ± 1.8) or control group (10.6 mm ± 2.0). In contrast, only the administration of TMZ showed a similar effectiveness than the previously observed for s.c. tumors, reducing significantly (p < 0.05) the growth
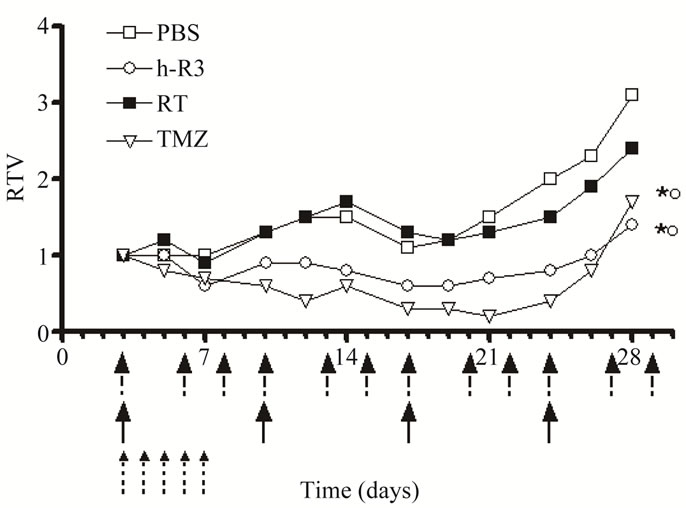
Figure 1. Therapeutic effect of radiation, or TMZ, or the anti-EGFR mAb nimotuzumab on the growth of established U87MG human GBM xenografted into NMRI nude mice. Tumor cells were inoculated into the flanks of nude mice. Treatments were initiated three days after tumor inoculation with ○ nimotuzumab (h-R3), 50 mg/kg intraperitoneally, three times/week by four weeks, or ■ radiation (RT), 4 Gy fractioned in 1 Gy weekly, or TMZ 10 mg/kg administered intraperitoneally on the first five consecutive days, or □ PBS control (PBS). Antibody administrations are showed as black arrows, radiation as fractioned arrows, and chemotherapy with TMZ with small fractioned arrows. Tumor volume was determined at the indicated time thereafter and RTV were calculated. Error bars are not shown because of better clarity. Kruskal-Wallis test; symbols indicate statistical differences as follows: *Significant to PBS; ºSignificant to radiation.
of intracranial (i.c.) tumors (1.2 mm ± 0.5). Moreover, non-statistical differences were found between group of mice treated with nimotuzumab or TMZ (p > 0.05).
Nevertheless, a histopathological analysis of i.c. tumor sections showed a strikingly different invasive growth pattern in mice treated either with nimotuzumab or the chemotherapeutic agent TMZ, in comparison to those maintained under radiation regimen (Figure 2(b)). Visualization under the microscope of i.c. tumor sections revealed that tumor surfaces and adjacent parenchyma in mice treated with radiation or controls were usually surrounded by numerous smaller satellite tumors, which is in contrast to tumors treated with nimotuzumab or TMZ. These results confirm our previous findings which suggest that nimotuzumab avoids the formation of microsatellite tumors induced by radiotherapy in this tumor model [11]. A further quantification of these satellites revealed that the satellite frequency (median, Min-Max) significantly decreased in mice treated with nimotuzumab (4, 0 - 8) or TMZ (1, 0 - 7) compared with radiation (21, 5 - 40) or control (19, 1 - 38) groups (Figure 2(c)).
All the U87MG-xenografted tumors were examined histologically after frozen tissue sectioning. A histologycal examination of these tumors showed no significant differences between specimens taken from different groups of treatments after therapy. Indeed, all tumors grew rapidly and were composed by fused cells displaying a fusiform shape; forming both transversal and longitudinal fiber bundles (representative images are shown in Figures 3(A)-(D)). Moreover, these histological findings were similar to those described for tumors arising from xenografts of the parental cell line U87MG [15].
To further evaluate mechanisms underlying the antitumor activity of each treatment, an immunohistochemical analysis was done at the end of the treatment in tumor specimens excised from the s.c. area. A positive EGFR immunostaining was detected in all analyzed tumor samples (Figures 3(E)-(H)). Data scored from +1 to +4 as per immunostaining intensity was blinded evaluated resulting nearly identical in each treatment group, indicating no differences in the EGFR expression level in analyzed tumors (Supplementary Figure 1).
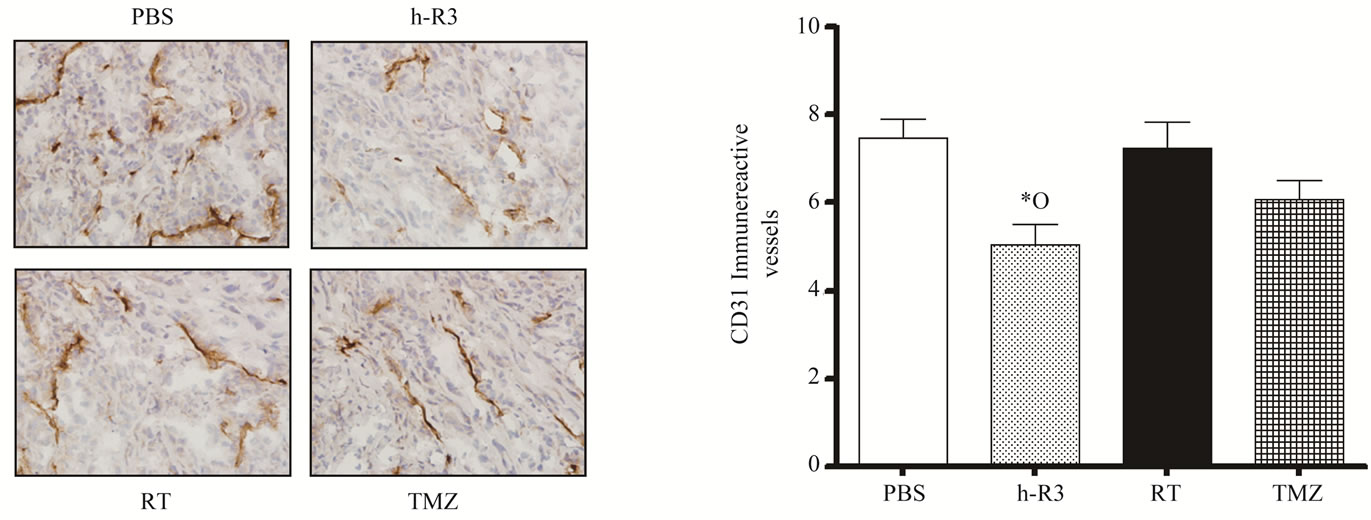
Earlier observations have postulated that malignant gliomas are highly dependent on angiogenesis for its growth and maintenance [16]. In addition, several drugs that act by blocking the EGFR (e.g. erlotinib, cetuximab and nimotuzumab) have shown an inhibitory effect on angiogenesis [11,17,18]. We therefore assessed the antiangiogenic activity of each therapy by analyzing tumor vessel density in s.c. tumors. A quantitative analysis of the blood vessels stained with the specific endothelial marker CD31 showed that only in nimotuzumab-treated mice, a significant decrease in the intratumoral microvessel density (MVD) was found. In contrast, in mice treated with TMZ only small vessel reductions were detectable, being not statistical different from those achieved in control or radiation groups (Figures 4(a)-(b)). Further, no differences were found between TMZ and radiation groups. These results confirmed the highly antiangiogenic potential of nimotuzumab in this tumor model [11].
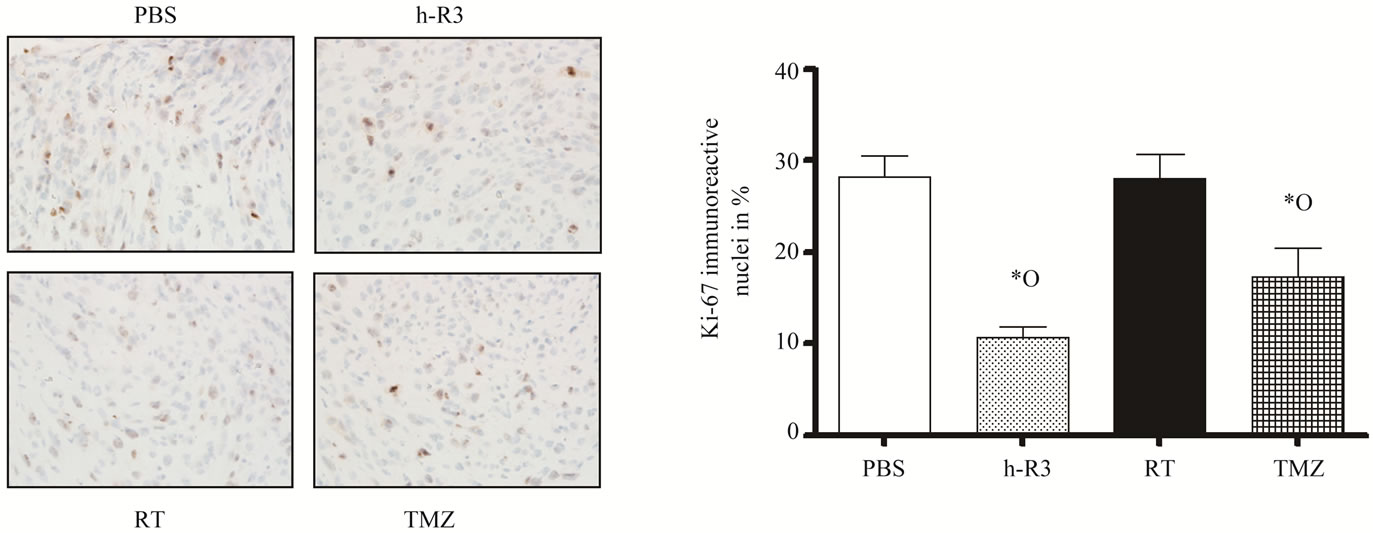
We next examined the inhibition of cell proliferation by analyzing the Ki-67 expression. The analysis of the proliferative activity in these tumors revealed that only nimotuzumab and TMZ significantly reduced the cell proliferation in treated mice, whereas radiation did not affect the proliferative activity of tumor cells, compared
 (a) (b) (c)
(a) (b) (c)
Figure 2. Therapeutic effect of radiation, or TMZ, or the anti-EGFR mAb nimotuzumab on the growth of established U87MG human GBM orthotopically xenografted into NMRI nude mice. (a) Tumor cells were injected intracranially in nude mice. Treatments were initiated three days after tumor inoculation with nimotuzumab (h-R3), 50 mg/kg intraperitoneally, three times per weeks by four weeks, or radiation (RT), 4 Gy fractioned in 1 Gy weekly, or TMZ 10 mg/kg administered intraperitoneally on the first five consecutive days, PBS control (PBS); (b) Mice were sacrificed and tumor volume was analyzed using a caliper. Stained sections show the extent and morphology of tumors treated with each therapy. The histologic tumor volume ± SEM is given. White arrowheads indicate tumors; (c) Analyzed brain sections from mice showed a remarkable reduction in the number of small satellite tumors in the groups of mice treated with nimotuzumab or TMZ, but not in those mice receiving RT. Kruskal-Wallis test; symbols indicate statistical differences as follows: *Significant to PBS; ºSignificant to radiation.
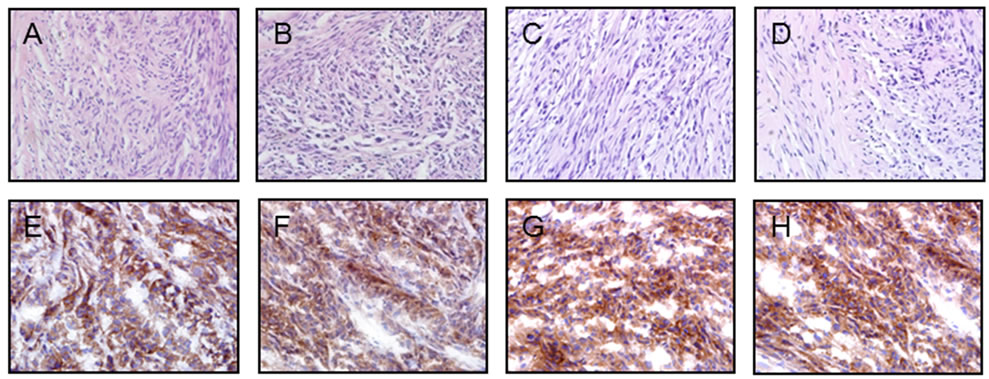
Figure 3. Morphology and immunohistochemistry of s.c. U87MG human GBM xenografts. Representative photomicrographs of tumors formed by implanting 107 U87MG cells into the flanks of mice and treated for 4 weeks with PBS (A and E), or nimotuzumab (B and F), or radiation (C and G), or TMZ (D and G). (A-D), HE; Tumors were composed by fused cells displaying a fusiform shape; forming both transversal and longitudinal fiber bundles; (E-H), Immunohistochemistry showed a strong positive pattern for EGFR expression with positive reaction in brown color revealed by DAB and counterstained with hematoxylin. Magnification 40×.
 (a) (b)
(a) (b)
Figure 4. Effect of radiation (RT), or TMZ, or nimotuzumab (h-R3) therapy on tumor blood vessel density. Immunostaining was performed using 5-µm tissue sections from s.c. tumors. (a) Micro vessels were detected by staining representative tumor sections with an antibody against CD31/PECAM-1. Subsequently, tumor blood vessels distribution was visualized under light microscopy; (b) Quantification of micro vessel density was assessed as described in “Materials and Methods”. Kruskal-Wallis test; symbols indicate statistical differences as follows: *Significant to PBS, ºSignificant to radiation. Magnification 40×.
to control. In the nimotuzumab group, the proliferative activity was reduced by 70% and 67% compared to control and radiation, respectively (Figures 5(a)-(b)). In TMZtreated tumors the proliferative activity was reduced by 49% compared to control, and no significant differences were found compared to nimotuzumab group.
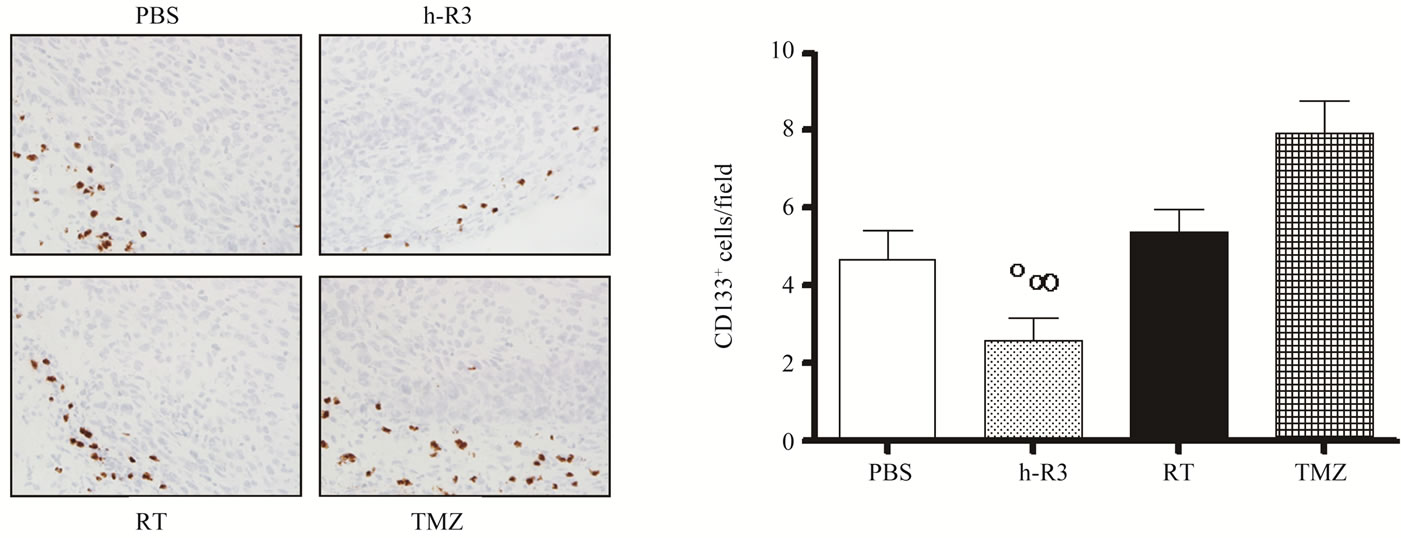
GBM are among the first solid cancers in which cancer stem cells (CSC) have been identified [19]. In these tumors the CD133+ but not CD133– cells have been reported to be responsible for tumor growth [19-21]. Furthermore, CSC population has shown to be a source of chemotherapy and radiation-therapy resistance within brain tumors [20,22,23]. We therefore investigated how different therapies may affect the frequency of CSC in U87MG xenografts, by quantifying the expression of the CD133 molecule in s.c. tumors. As shown in Figures 6(a)-(b), the administration of nimotuzumab resulted in significant reduction of CD133+ cells in tumor specimens. The frequency of CD133+ cells per field (mean ± sem) decreased from 4.7 ± 0.7 in control mice to 2.6 ± 0.6 in mice treated with nimotuzumab. As expected, the frequency of CD133+ CSC in the radiation group (5.4 ± 0.6), or in TZM group (7.9 ± 0.8) was slightly increased compared to controls, and significantly to nimotuzumab group, which is consistent with our previous observations [11]. These results are in line with previous findings demonstrating the resistance of CD133+ cells to the treatment with standard therapies including TMZ [22,23] and radiation [21]. In addition, these results corroborate the ability of antiEGFR monoclonal antibodies to target the CD133+ CSC
 (a) (b)
(a) (b)
Figure 5. Effect of radiation (RT), or TMZ, or nimotuzumab (h-R3) on tumor cell proliferation. (a) Immunostaining was performed using 5-µm tissue sections from s.c. tumors. Proliferating tumor cells were detected by staining representative tumor sections with the MIB-1 antibody. Subsequently, stained tumor sections were visualized on a light microscope; (b) Quantification of Ki-67-positive cells was assessed as described in “Materials and Methods”. Kruskal-Wallis test; symbols indicate statistical differences as follows: *Significant to PBS, ºsignificant to radiation. Magnification 40×.
 (a) (b)
(a) (b)
Figure 6. Effect of radiation (RT), or TMZ, or nimotuzumab (h-R3) on the CD133+ population. (a) Immunostaining was performed using 5-µm tissue sections from s.c. tumors. CD133+ cells were detected by staining representative tumor sections with the CD133/1 (AC133) antibody. Subsequently, stained tumor sections were visualized on a light microscope; (b) The frequency of CD133+ cells was quantified as described in “Materials and Methods”. Kruskal-Wallis test; symbols indicate statistical differences as follows: ºSignificant to radiation; ∞Significant to TMZ. Magnification 40×.
population in U87MG GBM tumors [11], being a potential therapeutic alternative to the failure of conventional therapies currently available for the treatment of brain tumors.
4. Discussion
GBM presents unique challenges to therapy due to its location, aggressive biological behavior and diffuse infiltrate growth [1]. Despite many technological advances in neurosurgery, neuroimaging, radiation therapy, and adjuvant chemotherapy, this malignancy remains one of the most lethal and challenging forms of cancer, with median survival time being approximately 14 months [2]. The disappointing results of standard therapy for GBM have led investigators to the search of new therapeutic options, such as therapies based on well-defined molecular targets.
One distinguishing feature of GBM is the high rate of amplification of the EGFR gene and the overexpression of the receptor. Precisely, high levels of EGFR expression have been largely correlated with aggressive behavior, increased metastasis, and decreased overall survival in GBM [24]. Recently, we examined nimotuzumab as a potent antitumor agent in the treatment of this malignancy in combination with radiotherapy, capable of inhibiting tumor cell growth of U87MG xenografts, acting as a modulator of radiation [11]. However, a study demonstrating the efficacy of nimotuzumab as a singleagent therapy in comparison to radiation or chemotherapy is mandatory for the introduction of this drug in the treatment of GBM. The reason for this is clear: radiation and chemotherapy (TMZ) are the “gold standards” for treatment of GBM and any novel therapy would be measured against them. Furthermore, such study would help us to elucidate the main mechanisms of antitumor activity of this drug as a useful starting point for the design of combination strategies. Here, we extend our previous observations to demonstrate that nimotuzumab can significantly inhibit the growth of U87MG tumor xenografts as single agent. A histological examination of xenografts suggests that these antitumor effects are exerted through a combination of antiangiogenic and antiproliferative actions. Using an orthotopic brain model, we further demonstrated that systemic administration of the antibody was less effective in inhibiting intracranial (i.c.) tumor growth, despite reducing the ability of tumor cells to migrate and invade the brain of mice.
The highly infiltrative nature of glioma cells is the major cause of dismal prognosis in malignant gliomas. For that reason, one of the therapeutic strategies to treat glioma is the eradication of invading glioma cell microsatellite tumors before they develop into recurrent tumors [25]. In line with this, several studies have showed that the blockade of the VEGF/VEGFR system by antiangiogenic and vascular targeting agents can inhibit the growth of i.c. gliomas [25]. However, there is also evidence that in different tumor models, antiangiogenic therapies can enhance tumor cell invasion or metastasis [26,27]. In the present study, the inhibitory effect of nimotuzumab on tumor growth of U87MG xenografts is accompanied by a reduction in tumor-induced angiogenesis, as assessed by CD31 immunostaining in tumor specimens (Figure 4). Moreover, in the i.c. model nimotuzumab significantly reduces the propensity of U87MG cells to migrate from the central core of implanted tumors (Figure 2(a)). In line with this, previous findings obtained from other tumor types revealed that the antiangiogenic activity of nimotuzumab is mediated by a potent downregulation of the VEGF in the squamous carcinoma cell line A431 in vitro as well as in vivo. Because VEGF acts as a survival factor for immature endothelial cells, protecting endothelial cells from apoptosis [28], a decreased production of VEGF induced by nimotuzumab may lead endothelial cell to apoptosis contributing to reduce neovascularity. Therefore, these results suggest that the inhibitory effect on vessel formation exerted by nimotuzumab might be mediated by an indirect action on endothelial cells via down-regulating angiogenic factors. Similar results have been reported for other anti-EGFR monoclonal antibodyies (e.g., cetuximab) and small-molecule tyrosine kinase inhibitors of the EGFR (e.g., gefitinib) [29]. Interestingly, the blockade of EGFR signaling has been reported to inhibit GBM cell migration and invasion [30,31]. Accordingly, the simultaneous blockade of EGFR and VEGFR has shown to inhibit tumor growth without increasing glioma cell invasion. In a study done by Lamszus et al., it was reported that inhibition of angiogenesis by DC101, a rat mAb against mouse VEGFR-2, can cause increased glioma cell invasion in an orthotopic human glioblastoma model [31]. In contrast, such increased tumor cell invasion was significantly inhibited by simultaneous blockade of EGFR with mAb cetuximab [31]. Consistent with these findings, we demonstrated that nimotuzumab partially abrogates the activation of the receptor, as well as the EGFR-downstream related signaling in U87MG tumor xenografts [11]. Altogether; these findings might partially explain why the antiangiogenic activity of nimotuzumab did not increase the tumor cell invasion in our experimental model. These observations suggest that nimotuzumab may act as an antiangiogenic and targeted therapy against both endothelial and tumor cells inhibiting tumor growth by disrupting the vascular endothelial microenvironment, in addition to abrogate the EGFR activation.
CSC has been identified as a major subpopulation of brain tumor cells with potent tumorigenic activity [32,33]. Increase evidences have demonstrated a close relationship between endothelial cells and brain CSC for the initiation and growth of tumors [34]. Previous studies suggest that CSC of orthotopic GBM xenografts may secrete angiogenic factors that promote the recruitment and formation of tumor blood vessels [21]. Moreover, the therapeutic activity of antiangiogenic therapies against GBM has been linked to their intrinsic capacity to deplete tumor blood vessels and CSC [34]. Here we confirmed previous findings demonstrating the ability of the anti-EGFR monoclonal antibodies to targeting the CD133+ CSC population in U87MG xenografts [11]. This may be achieved through a direct effect of the antibody on tumor cells or as a consequence of its antiangiogenic activity. A direct effect of nimotuzumab on the tumor cells is suggested by previous results indicating that nimotuzumab can decrease cell proliferation by inhibiting EGFR activation [35,36]. Interestingly, latest studies conducted by our group revealed that CSC markers Nestin and CD133 are co-expressed alongside the EGFR on the cell surface of the U87MG cells providing a plausible explanation for such findings (unpublished data). These results also support previous findings demonstrating that nimotuzumab efficacy is directly associated to the levels of EGFR expressed in the tumor cells [37]. Alternatively, an indirect targeting of CSC by nimotuzumab might also result from the disruption of the vascular microenvironment of tumors as a result of the antiangiogenic activity of nimotuzumab on tumor xenografts.
Unlike nimotuzumab, TMZ did not affect the CD133+ cells, despite inducing a significant antitumor activity. This may be explained by the low frequency of CD133+, which represents a minimal fraction of the U87MG tumor cells, despite its crucial role in the initiation, progression and recurrence of cancer [15]. TZM is an alkylating agent that exerts its cytotoxic effect mainly by methylating the DNA of replicating cells [38], leading to mismatched repairs and subsequent cell death [39]. Currently available data indicate that this adduct can be removed by the DNA repair protein O6-methylguanineDNA-methyltransferase (MGMT), which is expressed in a subgroup of GBM. Consequently TMZ displays its highest efficacy against tumors lacking MGMT expression; whereas the presence of the MGMT renders cells resistant to the cytotoxic actions of this agent [40]. In line with this, a higher expression of MGMT has been detected in the subgroup of CD133+ cells of GBM patients, which in turn show higher resistance to the treatment with TZM, compared to CD133– cells [23]. The ability of nimotuzumab to target the radio and chemoresistant CD- 133+ population not only contribute to a better understanding of nimotuzumab in GBM, but also bears implications for the design of future clinical investigations of the therapeutic efficacy of nimotuzumab in combination with standard cytotoxic therapies. It may also represent a new choice of treatment for those patients not amenable for receiving standard cytotoxic therapies. Further clinical trials examining the efficacy of nimotuzumab in a subgroup of GBM patients with MGMT expression and high levels of EGFR are currently in progress.
Our results also suggest a differential pharmacodynamic effect of nimotuzumab in tumors located in the flank or in the brains of xenografted mice. In subcutaneous (s.c.) tumors, nimotuzumab induced a significant increase in tumor growth delay. However, in brain tumors the antibody only produced a non-significant and modest reduction of tumor growth, despite inhibiting initial events in the metastatic processes, i.e., migration and local invasion of tumor cells. These findings support the notion that agents that block EGFR signaling may inhibit GBM cell invasion at much lower concentrations than are necessary for growth suppression [30]. The reduced efficacy of nimotuzumab in the i.c. setting might result from an impaired capacity for the antibody to overcome the blood-brain barrier (BBB). To show an optimal antitumor activity, antibodies injected systemically need to cross the BBB and have to reach the brain tumor in concentrations, which are sufficient to induce its therapeutic activity. Previous immunoscintigraphy studies with 99mTclabelled ior egf/r3, the predecessor murine version of nimotuzumab, outlined its ability to cross the BBB, showing a high sensitivity, specificity and accuracy for the in vivo detection of astrocytomas in patients [41]. Moreover, studies done after dual therapy with nimotuzumab plus radiation showed a positive antibody uptake by patients with GBM residual lesions, while subjects with complete responses showed no antibody accumulation at the previously known site of tumors [8]. However, both reports did not perform paired scintigraphy studies to compare the antibody uptake before and after therapy, and the EGFR expression was not evaluated in the tumor specimens before starting treatment [8]. Therefore, despite preliminary evidences suggest that nimotuzumab injected systemically is able to cross the BBB; it is possible that a significant amount of the initially injected antibody do not reach the brain in sufficient concentrations to induce a significant antitumor activity. This idea is further validated in our experimental conditions, where all therapies are evaluated in mice bearing both tumors simultaneously. This experimental model avoids variations associated to the technical handling of animals or any other source of variability inherent to the animal itself.
Even more interesting is the fact that tumors implanted in the flank of mice may sequester the free circulating drug, due to being located in an anatomical site more accessible for the antibody, avoiding higher accumulations of the drug in the i.c. tumors. This issue should not be pertinent to TMZ, a molecule of lower molecular weight and size compared to antibodies, which may partially explain why TMZ, unlike nimotuzumab, shows a similar efficacy in treating both s.c. and i.c. tumors.
One distinguishing feature of nimotuzumab is connected to its very favorable toxicity profile in comparison to other members of the same class of inhibitors (e.g., cetuximab or panitumumab) suggesting the possibility to use this antibody under long-term chronic treatment conditions [42,43]. Here we found that a four-week treatment with nimotuzumab significantly inhibited the cell proliferation, and subsequent U87MG tumor growth in s.c. tumors. These results contrast with previous findings in which nimotuzumab failed to exert a significant antitumor activity on U87MG tumors when the administration of the antibody was interrupted three weeks after the initiation of the treatment [11]. Therefore, these results reinforce the need of a sustained administration of the drug in the treatment of GBM. Based on these considerations, long-term chronic treatments with this drug should be next evaluated in the clinic.
5. Conclusion
We demonstrated that nimotuzumab, a monoclonal antibody against the EGFR, has efficacy in inhibiting the growth of U87MG xenografts as a single-agent. This antitumor activity is driven by a combination of a potent antiangiogenic activity and a reduction in tumor cell proliferation and invasiveness. In addition, its ability to effectively target the CD133+ radio and chemoresistant population may supports its use in the treatment of GBM in combination with conventional therapies. In view of these results, it might be recalled that nimotuzumab may represent a new option of treatment for those cancer patients not amenable for receiving standard cytotoxic therapies currently available in the management of GBM.
6. Acknowledgements
We thank Dr. Tania Aciego (Eusebio Hernandez Hospital) and Idael Pineda (Center of Molecular Immunology) for their technical assistance in preparing tumor samples for histological analysis and immunohistochemistry. We also acknowledge Monika Becker (Max Delbrück Center for Molecular Medicine) for assistance in animal experiments and Dr. Normando Iznaga (Center of Molecular Immunology) for technical discussions.
REFERENCES
- R. Stupp, W. P. Mason, M. J. van den Bent, M. Weller, B. Fisher, M. J. Taphoorn, K. Belanger, A. A. Brandes, C. Marosi, U. Bogdahn, J. Curschmann, R. C. Janzer, S. K. Ludwin, T. Gorlia, A. Allgeier, D. Lacombe, J. G. Cairncross, E. Eisenhauer and R. O. Mirimanoff, “Radiotherapy plus Concomitant and Adjuvant Temozolomide for Glioblastoma,” The New England Journal of Medicine, Vol. 352, No. 10, 2005, pp. 987-996. doi:10.1056/NEJMoa043330
- E. G. Van Meir, C. G. Hadjipanayis, A. D. Norden, H. K. Shu, P. Y. Wen and J. J. Olson, “Exciting New Advances in Neuro-Oncology: The Avenue to a Cure for Malignant Glioma,” CA: A Cancer Journal for Clinicians, Vol. 60, No. 3, 2010, pp. 166-193. doi:10.3322/caac.20069
- L. G. Feun, N. Savaraj and H. J. Landy, “Drug Resistance in Brain Tumors,” Journal of Neuro-Oncology, Vol. 20, No. 2, 1994, pp. 165-176. doi:10.1007/BF01052726
- J. N. Rich, “Cancer Stem Cells in Radiation Resistance,” Cancer Research, Vol. 67, No. 19, 2007, pp. 8980-8984. doi:10.1158/0008-5472.CAN-07-0895
- M. D. Prados and C. Russo, “Chemotherapy of Brain Tumors,” Seminars in Surgical Oncology, Vol. 14, No. 1, 1998, pp. 88-95. doi:10.1002/(SICI)1098-2388(199801/02)14:1<88::AID-SSU11>3.0.CO;2-5
- D. Beier, S. Rohrl, D. R. Pillai, S. Schwarz, L. A. KunzSchughart, P. Leukel, M. Proescholdt, A. Brawanski, U. Bogdahn, A. Trampe-Kieslich, B. Giebel, J. Wischhusen, G. Reifenberger, P. Hau and C. P. Beier, “Temozolomide Preferentially Depletes Cancer Stem Cells in Glioblastoma,” Cancer Research, Vol. 68, No. 14, 2008, pp. 5706- 5015. doi:10.1158/0008-5472.CAN-07-6878
- H. Hauch, M. Sajedi and J. E. Wolff, “Treatment Arms Summarizing Analysis of 220 High-Grade Glioma Studies,” Anticancer Research, Vol. 25, No. 5, 2005, pp. 3585-3590.
- T. C. Ramos, J. Figueredo, M. Catala, S. Gonzalez, J. C. Selva, T. M. Cruz, C. Toledo, S. Silva, Y. Pestano, M. Ramos, I. Leonard, O. Torres, P. Marinello, R. Perez and A. Lage, “Treatment of High-Grade Glioma Patients with the Humanized Anti-Epidermal Growth Factor Receptor (EGFR) Antibody h-R3: Report from a Phase I/II Trial,” Cancer Biology & Therapy, Vol. 5, No. 4, 2006, pp. 375-379. doi:10.4161/cbt.5.4.2522
- P. K. Julka, S. Mallick, T. Puri, S. Goyal, N. Joshi, N. Gupta and G. K. Rath, “Feasibility and Safety of Combining Nimotuzumab with Temozolomide and Radiotherapy in Adult Patients with Glioblastoma: An Indian Clinical Experience,” Journal of Clinical Oncology, Vol. 28, No. 15, 2010.
- U. Bode, S. Buchen, M. Warmuth-Metz, T. Pietsch, F. Bach and G. Fleischhack, “Final Report of a Phase II Trial of Nimotuzumab in the Treatment of Refractory and Relapsed High-Grade Gliomas in Children and Adolescents,” Journal of Clinical Oncology, Vol. 25, No. 18, 2007.
- A. Diaz Miqueli, J. Rolff, M. Lemm, I. Fichtner, R. Perez and E. Montero, “Radiosensitisation of U87MG Brain Tumours by Anti-Epidermal Growth Factor Receptor Monoclonal Antibodies,” British Journal of Cancer, Vol. 100, No. 6, 2009, pp. 950-958. doi:10.1038/sj.bjc.6604943
- C. Mateo, E. Moreno, K. Amour, J. Lombardero, W. Harris and R. Perez, “Humanization of a Mouse Monoclonal Antibody That Blocks the Epidermal Growth Factor Receptor: Recovery of Antagonistic Activity,” Immunotechnology, Vol. 3, No. 1, 1997, pp. 71-81. doi:10.1016/S1380-2933(97)00065-1
- “United Kingdom Co-ordinating Committee on Cancer Research (UKCCCR) Guidelines for the Welfare of Animals in Experimental Neoplasia (Second Edition),” British Journal of Cancer, Vol. 77, No. 1, 1998, pp. 1-10.
- A. M. Mansour, J. Drevs, N. Esser, F. M. Hamada, O. A. Badary, C. Unger, I. Fichtner, and F. Kratz, “A New Approach for the Treatment of Malignant Melanoma: Enhanced Antitumor Efficacy of an Albumin-Binding Doxorubicin Prodrug That Is Cleaved by Matrix Metalloproteinase 2,” Cancer Research, Vol. 63, No. 14, 2003, pp. 4062-4066.
- S. C. Yu, Y. F. Ping, L. Yi, Z. H. Zhou, J. H. Chen, X. H. Yao, L. Gao, J. M. Wang and X. W. Bian, “Isolation and Characterization of Cancer Stem Cells from a Human Glioblastoma Cell Line U87,” Cancer Letters, Vol. 265, No. 1, 2008, pp. 124-134. doi:10.1016/j.canlet.2008.02.010
- B. Kaur, F. W. Khwaja, E. A. Severson, S. L. Matheny, D. J. Brat and E. G. Van Meir, “Hypoxia and the HypoxiaInducible-Factor Pathway in Glioma Growth and Angiogenesis,” Journal of Neuro-Oncology, Vol. 7, No. 2, 2005, pp. 134-153. doi:10.1215/S1152851704001115
- T. Crombet-Ramos, J. Rak, R. Perez and A. Viloria-Petit, “Antiproliferative, Antiangiogenic and Proapoptotic Activity of h-R3: A Humanized Anti-EGFR Antibody,” International Journal of Cancer, Vol. 101, No. 6, 2002, pp. 567-575. doi:10.1002/ijc.10647
- J. Folkman, “Angiogenesis: An Organizing Principle for Drug Discovery,” Nature Reviews Drug Discovery, Vol. 6, No. 4, 2007, pp. 273-286. doi:10.1038/nrd2115
- S. K. Singh, C. Hawkins, I. D. Clarke, J. A. Squire, J. Bayani, T. Hide, R. M. Henkelman, M. D. Cusimano and P. B. Dirks, “Identification of Human Brain Tumour Initiating Cells,” Nature, Vol. 432, No. 7015, 2004, pp. 396- 401. doi:10.1038/nature03128
- S. Bao, Q. Wu, R. E. McLendon, Y. Hao, Q. Shi, A. B. Hjelmeland, M. W. Dewhirst, D. D. Bigner and J. N. Rich, “Glioma Stem Cells Promote Radioresistance by Preferential Activation of the DNA Damage Response,” Nature, Vol. 444, No. 7120, 2006, pp. 756-760. doi:10.1038/nature05236
- S. Bao, Q. Wu, S. Sathornsumetee, Y. Hao, Z. Li, A. B. Hjelmeland, Q. Shi, R. E. McLendon, D. D. Bigner and J. N. Rich, “Stem Cell-Like Glioma Cells Promote Tumor Angiogenesis through Vascular Endothelial Growth Factor,” Cancer Research, Vol. 66, No. 16, 2006, pp. 7843- 7848. doi:10.1158/0008-5472.CAN-06-1010
- A. Eramo, L. Ricci-Vitiani, A. Zeuner, R. Pallini, F. Lotti, G. Sette, E. Pilozzi, L. M. Larocca, C. Peschle and R. De Maria, “Chemotherapy Resistance of Glioblastoma Stem Cells,” Cell Death & Differentiation, Vol. 13, No. 7, 2006, pp. 1238-1241. doi:10.1038/sj.cdd.4401872
- G. Liu, X. Yuan, Z. Zeng, P. Tunici, H. Ng, I. R. Abdulkadir, L. Lu, D. Irvin, K. L. Black and J. S. Yu, “Analysis of Gene Expression and Chemoresistance of CD133+ Cancer Stem Cells in Glioblastoma,” Molecular Cancer, Vol. 5, 2006, p. 67. doi:10.1186/1476-4598-5-67
- N. Shinojima, K. Tada, S. Shiraishi, T. Kamiryo, M. Kochi, H. Nakamura, K. Makino, H. Saya, H. Hirano, J. Kuratsu, K. Oka, Y. Ishimaru and Y. Ushio, “Prognostic Value of Epidermal Growth Factor Receptor in Patients with Glioblastoma Multiforme,” Cancer Research, Vol. 63, No. 20, 2003, pp. 6962-6970.
- J. J. Vredenburgh, A. Desjardins, J. E. Herndon II, J. M. Dowell, D. A. Reardon, J. A. Quinn, J. N. Rich, S. Sathornsumetee, S. Gururangan, M. Wagner, D. D. Bigner, A. H. Friedman and H. S. Friedman, “Phase II Trial of Bevacizumab and Irinotecan in Recurrent Malignant Glioma,” Clinical Cancer Research, Vol. 13, No. 4, 2007, pp. 1253- 1259. doi:10.1158/1078-0432.CCR-06-2309
- E. S. Kim, A. Serur, J. Huang, C. A. Manley, K. W. McCrudden, J. S. Frischer, S. Z. Soffer, L. Ring, T. New, S. Zabski, J. S. Rudge, J. Holash, G. D. Yancopoulos, J. J. Kandel and D. J. Yamashiro, “Potent VEGF Blockade Causes Regression of Coopted Vessels in a Model of Neuroblastoma,” Proceedings of the National Academy of Sciences of the United States of America, Vol. 99, No. 17, 2002, pp. 11399-11404. doi:10.1073/pnas.172398399
- P. S. Steeg, “Angiogenesis Inhibitors: Motivators of Metastasis?” Nature Medicine, Vol. 9, No. 7, 2003, pp. 822- 823. doi:10.1038/nm0703-822
- G. P. Pidgeon, M. P. Barr, J. H. Harmey, D. A. Foley and D. J. Bouchier-Hayes, “Vascular Endothelial Growth Factor (VEGF) Upregulates BCL-2 and Inhibits Apoptosis in Human and Murine Mammary Adenocarcinoma Cells,” British Journal of Cancer, Vol. 85, No. 2, 2001, pp. 273- 278. doi:10.1054/bjoc.2001.1876
- L. M. Ellis, “Epidermal Growth Factor Receptor in Tumor Angiogenesis,” Hematology/Oncology Clinics of North America, Vol. 18, No. 5, 2004, pp. 1007-1021. doi:10.1016/j.hoc.2004.06.002
- P. L. Penar, S. Khoshyomn, A. Bhushan and T. R. Tritton, “Inhibition of Epidermal Growth Factor Receptor-Associated Tyrosine Kinase Blocks Glioblastoma Invasion of the Brain,” Neurosurgery, Vol. 40, No. 1, 1997, pp. 141- 151.
- K. Lamszus, M. A. Brockmann, C. Eckerich, P. Bohlen, C. May, U. Mangold, R. Fillbrandt and M. Westphal, “Inhibition of Glioblastoma Angiogenesis and Invasion by Combined Treatments Directed against Vascular Endothelial Growth Factor Receptor-2, Epidermal Growth Factor Receptor, and Vascular Endothelial-Cadherin,” Clinical Cancer Research, Vol. 11, No. 13, 2005, pp. 4934- 4940. doi:10.1158/1078-0432.CCR-04-2270
- H. D. Hemmati, I. Nakano, J. A. Lazareff, M. Masterman-Smith, D. H. Geschwind, M. Bronner-Fraser and H. I. Kornblum, “Cancerous Stem Cells Can Arise from Pediatric Brain Tumors,” Proceedings of the National Academy of Sciences of the United States of America, Vol. 100, No. 25, 2003, pp. 15178-15183. doi:10.1073/pnas.2036535100
- S. K. Singh, I. D. Clarke, M. Terasaki, V. E. Bonn, C. Hawkins, J. Squire and P. B. Dirks, “Identification of a Cancer Stem Cell in Human Brain Tumors,” Cancer Research, Vol. 63, No. 18, 2003, pp. 5821-5828.
- C. Calabrese, H. Poppleton, M. Kocak, T. L. Hogg, C. Fuller, B. Hamner, E. Y. Oh, M. W. Gaber, D. Finklestein, M. Allen, A. Frank, I. T. Bayazitov, S. S. Zakharenko, A. Gajjar, A. Davidoff and R. J. Gilbertson, “A Perivascular Niche for Brain Tumor Stem Cells,” Cancer Cell, Vol. 11, No. 1, 2007, pp. 69-82. doi:10.1016/j.ccr.2006.11.020
- A. Diaz Miqueli, R. Blanco, B. Garcia, T. Badia, A. E. Batista, R. Alonso and E. Montero, “Biological Activity in Vitro of Anti-Epidermal Growth Factor Receptor Monoclonal Antibodies with Different Affinities,” Hybridoma (Larchmt), Vol. 26, No. 6, 2007, pp. 423-431. doi:10.1089/hyb.2007.0516
- Y. Akashi, I. Okamoto, T. Iwasa, T. Yoshida, M. Suzuki, E. Hatashita, Y. Yamada, T. Satoh, M. Fukuoka, K. Ono and K. Nakagawa, “Enhancement of the Antitumor Activity of Ionising Radiation by Nimotuzumab, a Humanised Monoclonal Antibody to the Epidermal Growth Factor Receptor, in Non-Small Cell Lung Cancer Cell Lines of Differing Epidermal Growth Factor Receptor Status,” British Journal of Cancer, Vol. 98, No. 4, 2008, pp. 749- 755. doi:10.1038/sj.bjc.6604222
- M. O. Rodriguez, T. C. Rivero, R. C. Bahi, C. R. Muchuli, M. A. Bilbao, E. N. Vinageras, J. Alert, J. J. Galainena, E. Rodriguez, E. Gracias, B. Mulen, B. Wilkinson, E. L. de Armas, K. Perez, I. Pineda, M. Frometa, I. Leonard, V. Mullens, C. Viada, P. Luaces, O. Torres, N. Iznaga and T. Crombet, “Nimotuzumab plus Radiotherapy for Unresectable Squamous-Cell Carcinoma of the Head and Neck,” Cancer Biology & Therapy, Vol. 9, No. 5, 2010, pp. 343-349. doi:10.4161/cbt.9.5.10981
- B. J. Denny, R. T. Wheelhouse, M. F. Stevens, L. L. Tsang and J. A. Slack, “NMR and Molecular Modeling Investigation of the Mechanism of Activation of the Antitumor Drug Temozolomide and Its Interaction with DNA,” Biochemistry, Vol. 33, No. 31, 1994, pp. 9045-9051. doi:10.1021/bi00197a003
- S. D’Atri, L. Tentori, P. M. Lacal, G. Graziani, E. Pagani, E. Benincasa, G. Zambruno, E. Bonmassar and J. Jiricny, “Involvement of the Mismatch Repair System in Temozolomide-Induced Apoptosis,” Molecular Pharmacology, Vol. 54, No. 2, 1998, pp. 334-341.
- M. E. Hegi, A. C. Diserens, T. Gorlia, M. F. Hamou, N. de Tribolet, M. Weller, J. M. Kros, J. A. Hainfellner, W. Mason, L. Mariani, J. E. Bromberg, P. Hau, R. O. Mirimanoff, J. G. Cairncross, R. C. Janzer and R. Stupp, “MGMT Gene Silencing and Benefit from Temozolomide in Glioblastoma,” The New England Journal of Medicine, Vol. 352, No. 10, 2005, pp. 997-1003. doi:10.1056/NEJMoa043331
- M. Ramos-Suzarte, N. Rodriguez, J. P. Oliva, N. Iznaga-Escobar, A. Perera, A. Morales, N. Gonzalez, M. Cordero, L. Torres, G. Pimentel, M. Borron, J. Gonzalez, O. Torres, T. Rodriguez and R. Perez, “99mTc-Labeled Antihuman Epidermal Growth Factor Receptor Antibody in Patients with Tumors of Epithelial Origin: Part III. Clinical Trials Safety and Diagnostic Efficacy,” Journal of Nuclear Medicine, Vol. 40, No. 5, 1999, pp. 768-775.
- W. K. Boland and G. Bebb, “Nimotuzumab: A Novel Anti-EGFR Monoclonal Antibody That Retains AntiEGFR Activity While Minimizing Skin Toxicity,” Expert Opinion on Biological Therapy, Vol. 9, No. 9, 2009, pp. 1199-1206. doi:10.1517/14712590903110709
- G. Bebb, W. Boland and B. Melosky, “Don’t Jump to Rash Conclusions,” Cancer Biology & Therapy, Vol. 11, No. 7, 2011, pp. 639-641. doi:10.4161/cbt.11.7.14920
Abbreviations
BBB: blood-brain barrier CSC: cancer stem cells EGF: epidermal growth factor EGFR: EGF receptor GBM: glioblastoma multiforme mAb: monoclonal antibody MVD: microvessel density TMZ: temozolomide VEGF: vascular endothelial growth factor VEGFR: VEGF receptor
NOTES
*Corresponding author.