Journal of Cancer Therapy
Vol.1 No.2(2010), Article ID:2033,11 pages DOI:10.4236/jct.2010.12013
Mechanisms and Immune Dysregulation in Arsenic Skin Carcinogenesis
![]()
1Department of Dermatology, Kaohsiung Medical University Hospital and Kaohsiung Medical University College of Medicine, Kaohsiung, Taiwan, China; 2Department of Dermatology, Kaohsiung Municipal Hsiao-Kang Hospital, Kaohsiung, Taiwan, China.
Email: dermyu@kmu.edu.tw
Received April 26th, 2010; revised May 19th, 2010; accepted May 25th, 2010.
Keywords: Arsenic, Bowen’s disease, Skin cancer, Innate immunity, Langerhans cells
ABSTRACT
Long-term exposure to arsenic is associated with cancers of lung, urinary bladder, kidney, liver and skin. Arsenic carcinogenesis might result from oxidative stress, altered growth factors, chromosomal abnormality, immune dysregulation, and aberrant epigenetic regulations. Bowen’s disease (As-BD) is the most common form of arsenic-induces skin cancers and is characterized by chronicity, multiplicity, and predisposition in sun-spare skin. However, only about 1% of the population exposed to arsenic developped skin cancers, indicating the host immune response plays an important modulatory role in skin carcinogenesis. In this review, we review the pathomechanisms of arsenic skin carcinogenesis and the immune interactions. Arsenic affects innate and adaptive immune responses through CD4+ T cells, monocytes, macrophages, and Langerhans cells. In skin of As-BD, CD4+ T cells undergo selective and differential apoptosis via Fas-FasL interaction. Numbers and dendrites of Langerhans cells are reduced in As-BD lesions. There is a defective homeostasis and aberrant trafficking of Langerhans cells. Such information is essential to understand the molecular mechanism for arsenic carcinogenesis in both skin and in internal organs.
1. Introduction
Arsenic is a ubiquitous element on the earth. People may expose to arsenic in several ways through drinking, inhalation, and direct skin contact. Drinking of water with arsenic remains the major route of human exposure [1], leading to development of cancers of skin, lungs, and liver in many countries, including Bangladesh, Taiwan, West Bengal of India, Chile, Mexico, and China [2]. Industrially, arsenic is used to generate paints and insecticides. Arsenide is also a critical constituent in semiconductors that is used for electronic chips and computers. In addition to its carcinogenic property, arsenic exposure is also associated with vascular diseases, including stroke, ischemic heart diseases, and peripheral vascular disease [3]. In contrast to its notoriously adverse health effects, arsenic has been used for treatment of lymphoma and leukemia and it still remains the drug of choice for acute promyelocytic leukemia.
It is estimated that around 10% of population exposed to arsenic will develop skin abnormalities, including variegated hyper-/hypo-pigmentations, arsenic keratosis, Bowen’s disease, and invasive skin cancers. Only about 1% of exposed population develops skin cancers. Longterm arsenic exposure results in impaired immunity in susceptible individuals, which may account for the development of cancers in vulnerable individuals. Personal genetic variability, immune system, and the interaction of both might differ, leading to differential susceptibility and cancer immunity in the process of arsenic carcinogenesis.
Based on its oxidative status, arsenic exists in two inorganic chemical forms. Arsenite (AsIII) is a trivalent form while arsenate (AsV) is a pentavalent form. Trivalent arsenite is about 2-10 times more toxic than pentavalent arsenate. In tissues, arsenic is methylated by methyl group supplied by s-adenosylmethionine (SAM). Compared to inorganic forms, the methylated metabolites are less genotoxic [4] and are excreted more quickly in urine [5,6]. After ingestion, inorganic arsenic is obtained by erythrocytes and then distributed systemically to multiple organs, including lungs, liver, and skin [7,8]. In transit from blood to tissues, arsenate is reduced to arsenite. In the liver, arsenic is methylated into monomethylarsenic acid (MMA V), which can be further reduced to monomethyl arsonous acid (MMA III). A further methylation reaction modifies MMA III to dimethylarsinic acid (DMA V) [9]. In the process of methylation process, several reactive oxygen species (ROS) are generated [10,11]. WHO recommends safe groundwater arsenic concentration up to 50 μg/L but proposes a provisional arsenic standard at 10 μg/L [12]. The government regulation for arsenic contents in drinking water depends on countries but the standard generally is more stringent, for example, up to 10 g/L in Taiwan, Japan, and U.S.
One of the most systematized epidemiological studies for health effects of arsenic is conducted in Taiwan and it has led the basis of many epidemiological risk assessments over the last 40 years worldwide [6]. In the past decades, we have been investigating on the arsenic carcinogenesis with particular focus on skin not only it is readily accessible but also it might provide a model of chemical carcinogenesis and immune interaction. This review discusses the pathomechanisms of arsenic skin carcinogenesis with special focus on the interactions of immune system and arsenic-induced cancers.
2. Proposed Mechanisms of Action in Arsenical Carcinogenesis
Although arsenic is documented as a weak mutagen, the International Agency for Research on Cancer (IARC) has categorized arsenic as a human carcinogen [13]. The mechanism of arsenic carcinogenesis remains uncertain. However, oxidative stress, chromosomal abnormality and altered growth factors may contribute to arsenic carcinogenesis [9,14,15]. It has been suggested that arsenic might act as a co-carcinogen or a promoter in carcinogenesis by mode of action studies [16]. However, recent studies showed perinatal maternal exposure to arsenic results in spontaneous cancer developments in off springs, suggesting that arsenic might also act in the initiation in two stage chemical carcinogenesis [17]. The effects of arsenic exposure in early life development include epigenetic effects, via DNA hypomethylation, endocrine effects (most classes of steroid hormones), immune suppression, neurotoxicity, and interaction with enzymes critical for fetal development and programming [18].
Arsenic tends to bind to the thio-group (-SH) of proteins, targeting regulatory or structural proteins [19,20]. Approximately 200 proteins could be targeted by the bindings and interactions of arsenic-thio group [21]. Among these proteins, the proto-oncogene c-Jun is well investigated. By binding to thio-groups, arsenic can block Jun N-terminal kinase (JNK) phosphatase activity, resulting in an over activation of JNK, which activates proto-oncogene c-Jun, inducing c-Jun/c-Fos (AP-1)mediated gene upregulations [22-24]. These upregulated genes include cell cycle regulation, and apoptotic signaling, all of which are strongly linked to arsenic carcinogenesis. Moreover, we have shown there is a quantitative impairment of phosphorylation of keratin 1 and keratin 2 in the process of chronic arsenism, suggesting that keratins, containing plenty of thio groups, are the cellular targets of arsenic [25]. Through oxidative stress induced by arsenic, genomic mutations might develop, leading to initiation in carcinogenesis. Several lines of compelling evidences revealed the oxidative DNA damages in the target organ of arsenic-exposed animals and humans. In fact, clinical studies in arsenic-induced Bowen’s disease (As-BD) uncovered the correlations between increased 8-OHdG levels and the arsenic concentration in the lesional skin [26], indicating the importance of oxidative stress in arsenical skin carcinogenesis. Mechanistically, in vitro studies showed low concentrations of arsenic (< 5 µM) can generate ROS, which in term increases the transcription of the nuclear factor kappa B (NF-KB) [24,27-30], that eventually promotes cell proliferation [31].
The second possible mechanism in arsenic carcinogenesis is through genomic instability and chromosome abnormalities. Arsenic is repeatedly reported to induce chromosome abnormalities and aberrant sister chromatid exchanges [32-34]. In human fibroblasts and CHO cells, arsenic induced chromosome abnormalities and induces sister chromatid exchanges at high and low concentrations, respectively [35,36]. These chromosomal abnormalities were highly associated with arsenic-induced oxidative DNA damages [26,37] and might link to arsenic carcinogenesis. Arsenite exposure induced micronuclei (MN) formation [38], which indicates cellular response to DNA damages. An increased frequency of MN was also detected in exfoliated bladder cells, buccal cells, and lymphocytes from arsenic-exposed humans [39-41]. Chien et al showed that arsenite results in tumorigenicity of HaCaT cells in nude mice by increased frequency of MN [42].
The third possible etiological factor leading to arsenic carcinogenesis is through abnormal DNA repair and epigentic regulations. Arsenic was able to inhibit DNA repair systems in the steps of nucleotide excision repair [43-45], DNA ligase III activity, DNA base excision repair [46,47] and DNA strand break rejoining [48]. Many key DNA repair regulatory proteins were inhibited by arsenic, including DNA ligase I, DNA ligase II, DNA ligase III, DNA polymerase β, 6-methyl-guanine-DNA methyltransferase, and poly (ADP-ribose) polymerase (PARP) [14,47,49]. Agents messing up with those DNA repair proteins can lead to genetic mutations. Along with its effect in DNA repair, arsenic also potentiated the mutagenicity of other carcinogeneic factors (such as UV, X-rays, and chemical agent) [50-53]. Moreover, arsenic affected global histone methylation and also DNA methylation, indicating that arsenic also affects epigenome machinery to influence gene expressions involved in carcinogenesis [54]. In most cases, arsenic induced DNA hypomethylation, probably through the inhibition of DNA methyltransferases [55]. However, it has been reported there is a hypermethylation in promoter of gene p53 and p16 in people exposed to arsenic [56]. Arsenic at very low concentrations (below 1mM) can inhibit both DNMT1 and DNMT3A in HaCaT cells [57]. Indeed, recent studies showed perinatal exposure to arsenic results in DNA methylation globally in GC-rich (guanidine and cysteine) regions [58] .
3. Hophysiology of Arsenic Skin Cancers
Arsenic tends to accumulate in ectodermal tissues including the skin, hair and nails. Skin lesions are most common and most accessible in arsenic-induced pathologies [59-61]. Variegated hyperand hypo-pigmentation and punctate palmar-plantar hyperkeratosis are all hallmarks of chronic arsenic exposure. The hyperkeratosis may appear as a regular thickening or as discrete nodules. A dose relationship has been found for the arsenic concentrations in well water and the occurrences of hyperpigmentation and hyperkeratosis among the people living in the endemic areas [62,63]. Furthermore, Tseng et al. found a dose-response relationship between arsenic levels in drinking water and skin cancers by a comprehensive epidemiological survey five decades ago [62]. Among skin cancers, the most common arsenic-induced skin cancers are Bowen’s disease, followed by basal cell carcinoma and squamous cell carcinoma [62].
Bowen’s disease is a carcinoma in situ of the skin resulting from UV or arsenical exposure [2,59,62,64]. Clinically, arsenic-induced Bowen’s disease is different from classical (UV-induced) Bowen’s disease by the its multiplicity and its propensity in sun-spare skins [2,62,65]. There are abnormal cellular proliferation and apoptosis in arsenic-induced Bowen’s disease (As-BD) as presented with increased epidermal thickness and individual dyskeratotic keratinocytes, respectively [2,59]. After decades of development, As-BD can penetrate through basement membrane and become invasive squamous cell carcinoma (SCC), basal cell carcinoma (BCC), and combined forms of the skin cancer [2,62, 66,67]. Individuals with As-BD are considered a risk for development of malignancies in the lung and urinary bladder [67-70]. It was estimated that As-BD started within one decade, invasive skin cancer after scores of years [71], and lung cancers after 30 years following the chronic arsenic exposure [66]. Therefore, the characteristic clinical and histopathological features of As-BD serve as a model to understand the different stages of chemical carcinogenesis.
Microscopically, p53 protein was greatly expressed in As-BD as compared with non-arsenical BD [72-74]. Arsenic can induce p53 accumulation via an ATM-dependent pathway [75,76]. The over-expressed p53 in As-BD lesions was a mutant form [77,78], of which most of the p53 mutation sites are located on exon 5 and exon 8. Furthermore, the mutation types of p53 gene mutation in As-BD were different from those in UV-related skin cancers [79], indicating the differences in the pathogenesis of As-BD and UV-induced Bowen’s disease. Although the connection between p53 mutation and arsenic exposure was not clear, the effect of arsenic on p53-related pathways was well recognized. Studies have shown that arsenic exposure resulted in G2/M cell cycle arrest and DNA aneuploidy, both of which are regulated by p53 [80-82].
There are coexisting hyperproliferative and dyskeratotic (apoptotic) keratinocytes in As-BD lesions [31]. The effects of arsenic on keratinocytes depend on the concentrations of arsenic. At lower concentrations (≦ 1 μM), arsenic induced keratinocyte proliferation and enhanced both NF-КB and AP-1 activity [31]. The proliferation is dependent on the mitochondrial biogenesis (manuscript in preparation). At higher concentrations (≧ 5 μM), arsenic induced keratinocyte apoptosis by Fas/Fas ligand (FasL) pathway. Since promoter regions of FasL contained binding sites for AP-1, arsenic-activated Fas/ FasL signaling may associate with arsenic-induced AP-1 activation [83-85].
4. Arsenic Influences Immune Regulation and Immune Responses
Intact and functional immunity is important in tumor surveillance of skin cancers. This is evidenced by the fact that patients with renal transplant and HIV infection have higher risk to develop skin cancers. Depends on cell type, tissue, and species, arsenic influenced immune system and its responses differentially in many aspects [86-88]. Arsenic may provoke immune responses by inhibition of regulatory or suppressor cells. For example, arsenic affected function of regulatory T lymphocytes in humans [89]. Arsenic in vitro enhanced immune response by deleting the precursors of suppressor T cells from normal spleen cells [90].
Arsenic affected many different kinds of cells in the immune system, leading to dysregulated immune responses. In utero exposure to arsenic impaired child thymic development and enhanced morbidity to respiratory infection [91]. The increase in respiratory infection by influenza was also shown in arsenic-exposed mice [92] and was associated with alternation of immune response genes in the lungs, such as IL-1beta, IL-1R and a number of toll-like receptors [93]. In addition to its adverse effects on the infection, arsenic can alter the systemic immunity. In patients with As-BD, there was a markedly reduced in contact hypersensitivity (CHS) reaction to DNCB in the skin [94], accompanied with reduced spontaneous and phytohemaglutinin(PHA)-induced IFNgamma and TNF-alpha productions. The decreased CHS response to DNFB was also shown in the mice fed with arsenic-containing water [95]. We reported that arsenic induces CD4+ cells apoptosis by affecting the autocrine TNF-alpha loop [96,97]. Furthermore, there was a decrease in numbers of T cells and the expression of IL-2R on them from patients with As-BD [94,98]. We also showed that T cells from arsenic-exposed people were anergy to PHA stimulation but were exaggerative to arsenic treatment [96]. A recent study showed T cell proliferation to Concanavalin A (Con A) was markedly reduced in people exposed to arsenic and there was a parallel decrease in the levels of TNF-alpha, IFN-gamma, IL-2, IL-10, IL-5, and IL-4 [99]. In arsenic-exposed children, arsenic burden was also associated with a reduced proliferative response to PHA stimulation. CD4+ cells were selectively decreased with CD8+, B, and NK cells remained unaffected in proportion. IL-2 but not IL-4, IL-10, or IFN-gamma was decreased in PHA-activated peripheral blood mononuclear cells [100]. In mice, T cell-dependent humoral immune response was extremely sensitive to suppression by arsenic and assessment of humoral immune responses should be considered in evaluating the health effects of arsenic containing agents [101]. Collectively, arsenic inhibits systemic T cell activation and proliferation via TNF-alpha axis. Macrophages were also potential targets of arsenic in humans [102]. Macrophages from people exposed to arsenic showed loss of cell adhesion capacity, decreased in NO production, and impaired phagocytic ability [103]. Monocytes from children exposed to arsenic produced less superoxide anion and nitric oxide [104]. Arsenic also affected phagocytic ability of neutrophils and degranulation via Syk activation [105].
In the cell level, arsenic has dual effects on cell proliferation in lymphocytes. Arsenic compounds at low concentrations enhanced DNA synthesis in PHA-stimulated proliferation of human lymphocytes, whereas arsenic at higher concentrations inhibited cellular proliferation and induced apoptosis [96,106,107]. As occurred in fibroblasts and CHO cells, arsenic has been shown to induce aneuploidy [108,109], sister chromatid changes [110-112], other chromosomal abnormalities [32] in lymphocytes. Mechanistically, arsenic influenced T cell receptor activation by increasing basal and induced phosphorylation of Lck and Fyn (first kinase associated to TCR complex) [113]. Metabolites of arsenic can also interfere with cell division via tubulin disruption, leading to aneuploidy [114]. A major cellular source of methyl group, S-adenosyl-L-methionine can reverse micronucleus formation induced by sodium arsenite and other cytoskeleton disrupting agents in cultured human lymphocytes [115].
In contrast to the scarce studies studying effects of arsenic on normal lymphocytes, there are a lot of studies investigating arsenic induced apoptosis in cells from lymphoma or leukemia, which coincides with the use of arsenic trioxide in acute promyelocytic anemia and multiple myeloma. Arsenic induced normalization of differentiation of promyelocytes in acute promyelocytic leukemia, of which arsenic remains the drug of choice. Phosphoinositide 3-kinase/Akt inhibition increased arsenic trioxide-induced apoptosis of acute promyelocytic and T-cell leukemia [116]. Arsenic induced apoptosis through activation of Bax in hematopoietic cells [117]. Furthermore, arsenic trioxide (As) and interferon (IFN)alpha coordinately induced cell cycle arrest and apoptosis that is modulated by bcl-2, bax, p53, and NF-kappaB [118,119]. Thus, the effects of arsenic on T cells development may act as a double-sided sword and appear to be cell-specific and concentration-dependent.
5. Altered Skin Associated Lymphoid Tissue in Arsenic-Induced Bowen’s Disease
In patients with As-BD, there was a markedly reduced in contact hypersensitivity reaction to DNCB in the skin [94], accompanied with reduced spontaneous and PHAinduced IFN-gamma as well as TNF-alpha production. Both GM-CSF and TGF-alpha were found in the epidermis at clinically normal sites within 10 weeks after arsenic treatment in vivo and also from arsenic-treated keratinocytes [120,121]. Arsenic can enhance keratinocytes to express TGF-α, GM-CSF, IL-6 and IL-8 [120- 122]. These growth factors and cytokines expression may induce cutaneous tumorigenesis via AP-1 and NF-КB regulation [123] . Although a clear link has been established for impaired T cell proliferation by arsenic, the causative role of impaired T cell activation and predilection of T cell apoptosis with the cutaneous carcinogenesis has seldom discussed. There was a decreased proportion of peripheral CD4+ cells in the peripheral blood from arsenic-exposed humans as compared to that from control subjects; the CD4+ cells from As-BD patients were less vulnerable to arsenic-induced apoptosis, due to defected TNF-R1 expression. Those residual CD4+ cells were less susceptible to arsenic-induced apoptosis. However, when those CD4+ cells infiltrated into the As-BD lesions, FasL from As-BD in the epidermis induced selective CD4+ cell apoptosis (Figure 1). This additional tumor evasion phenomenon present in the cutaneous environment provided a reasonable explanation for persisting nature of arsenic cancers in the skin despite the moderate dermal inflammatory infiltrates [124].
It is evident that cell-mediated immunity is depressed in patients with arsenic-induced Bowen’s disease [94]. Langerhans cells (LC) are known to be one of the professional antigen presenting cells for T lymphocytes. They play a pivotal role in the presentation of tumor-associated antigens. Others and we have reported there was a progressive decrease in numbers of Langerhans cell in the order of normal skin, normal appearing skin in As-BD, and As-BD lesion. [125-127]. However, how arsenic alters LC migration and polarizes Th responses remains unknown. Using an epicutaneous protein sensitization model in mice, we have found that arsenic exposure enhanced LC migration to draining lymph nodes, and
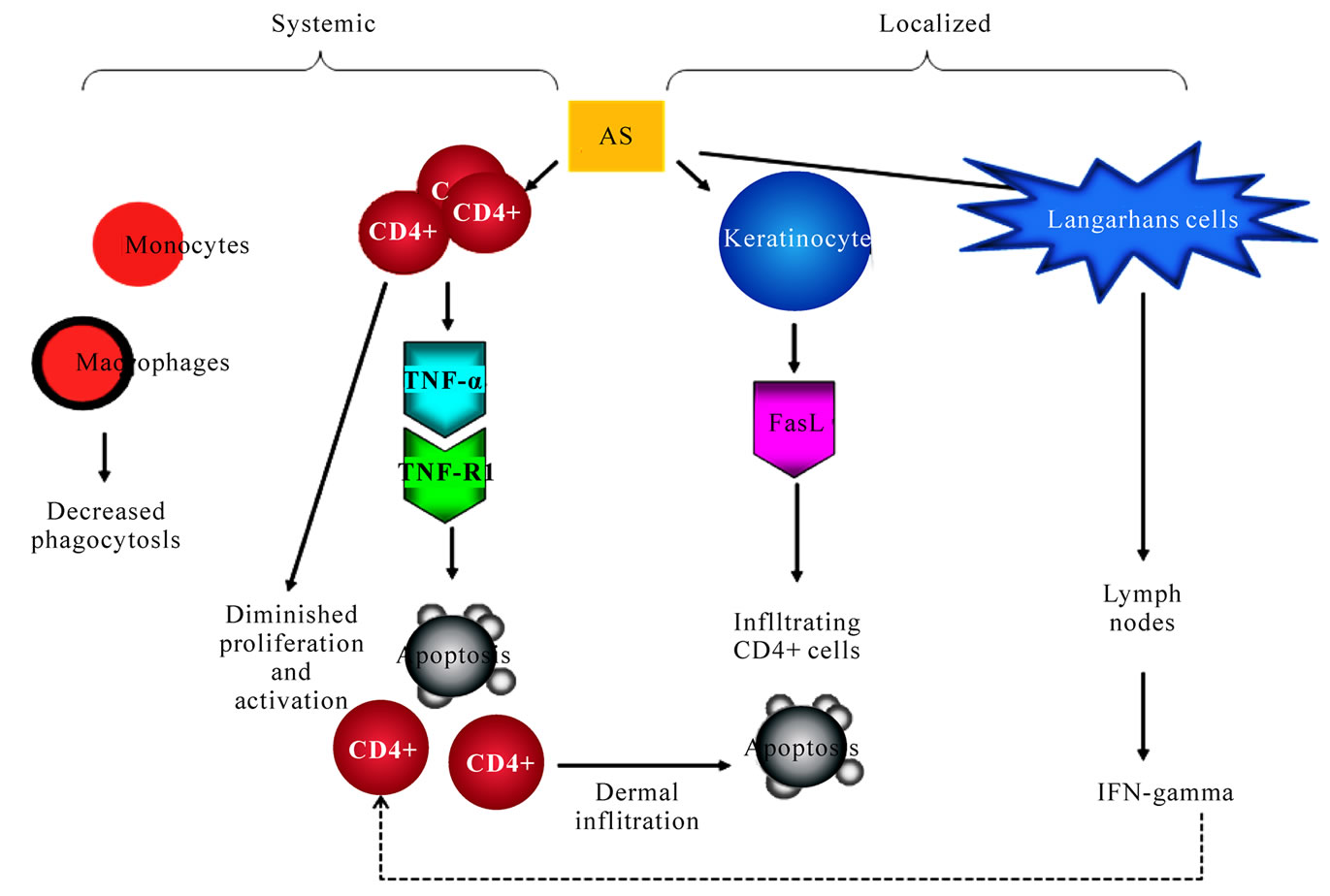
Figure 1. Proposed altered systemic and cutaneous immune responses in arsenic carcinogenesis. Arsenic inhibits T cell proliferation, induces T cell apoptosis, decreases monocyte/macrophage phagocytosis in the systemic circulation. In the skin, CD4+ T cells are vulnerable to apoptosis by FasL from epidermal keratinocytes. Langerhans cell is decreased in numbers in the epidermis and its trafficking to lymph nodes is enhanced
that enhanced LC migration to draining lymph nodes, and that the chronic nature of As-BD might result from enhanced Th1 responses with dysregulated LC trafficking (manuscript in preparation).
6. Conclusions
Bowen's disease is the most common form of arsenic-induced skin cancers. As-BD is characterized by its chronicity, multiplicity and predisposition in sun-spare skin. Patients with As-BD are often defected in their cellular immunity (Figure 1). CD4+ T cells is quantitatively reduced in people with As-BD. CD4+ T cells from arsenic-exposed individuals are less susceptible to apoptosis due to an impaired TNF-alpha-TNFR loop. However, once CD4+ T cells gain access the As-BD lesions, FasL from epidermal keratinocytes in As-BD induces selective CD4+ cell apoptosis. Notably, there is also a decrease in numbers and aberrant trafficking of Langerhans cells in As-BD. Thus, arsenic differentially affects systemic and localized immunity in arsenic skin carcinogenesis. In chemical carcinogenesis, the attribute of chemical immunology should be considered.
7. Acknowledgements
This study was supported by the National Science Council research grant NSC93-2320-B-002-059 and the National Health Research Institute research grant NHRIEX94-9231SI, Taiwan.
REFERENCES
- T. Gebel, “Confounding Variables in the Environmental Toxicology of Arsenic,” Toxicology, vol. 144, no. 1-3, 3 April 2000, pp. 155-162.
- H. S. Yu, C. H. Lee, S. H. Jee, C. K. Ho and Y. L. Guo, “Environmental and Occupational Skin Diseases in Taiwan,” Journal of Dermatology, vol. 28, no. 11, November 2001, pp. 628-631.
- J. C. States, S. Srivastava, Y. Chen and A. Barchowsky, “Arsenic and Cardiovascular Disease,” Toxicology Science, vol. 107, no. 2, February 2009, pp. 312-323.
- M. M. Moore, K. Harrington-Brock and C. L. Doerr, “Relative Genotoxic Potency of Arsenic and its Methylated Metabolites,” Mutation Research, vol. 386, no. 3, June 1997, pp. 279-290.
- J. P. Buchet, R. Lauwerys and H. Roels, “Urinary Excretion of Inorganic Arsenic and its Metabolites after Repeated Ingestion of Sodium Metaarsenite by Volunteers,” International Archives of Occupational and Environmental Health, vol. 48, no. 2, 1981, pp. 111-118.
- Y. K. Huang, Y. L. Huang, Y. M. Hsueh, J. T. Wang, M. H. Yang and C. J. Chen, “Changes in Urinary Arsenic Methylation Profiles in a 15-Year Interval after Cessation of Arsenic Ingestion in Southwest Taiwan,” Environmental Health Perspectives, vol. 117, no. 12, December 2009, pp. 1860-1866.
- H. Yamauch and B. A. Fowler, “Toxicity and Metabolism of Inorganic and Methylated Arsenicals,” In: J. Q. Nriagu Ed., Arsenic in the Environment, Part II, Human Health and Ecosystem Effects, John Wiley and Sons, New York, 1994, pp. 35-53.
- Y. M. Hsueh, Y. L. Huang, C. C. Huang, W. L. Wu, H. M. Chen, M. H. Yang, L. C. Lue and C. J. Chen, “Urinary Levels of Inorganic and Organic Arsenic Metabolites among Residents in an Arseniasis-Hyperendemic Area in Taiwan,” Journal of Toxicology and Environmental Health A, vol. 54, no. 6, 24 July 1998, pp. 431-444.
- C. Kojima, D. C. Ramirez, E. J. Tokar, S. Himeno, Z. Drobna, M. Styblo, R. P. Mason and M. P. Waalkes, “Requirement of Arsenic Biomethylation for Oxidative DNA Damage,” Journal of the National Cancer Institute, vol. 101, no. 24, 16 December 2009, pp. 1670-1681.
- K. Yamanaka, M. Hoshino, M. Okamoto, R. Sawamura, A. Hasegawa and S. Okada, “Induction of DNA Damage by Dimethylarsine, a Metabolite of Inorganic Arsenics, is for the Major Part Likely Due to its Peroxyl Radical,” Biochemical and Biophysical Research Communications, vol. 168, no. 1, 16 April 1990, pp. 58-64.
- M. J. Mass, A. Tennant, B. C. Roop, W. R. Cullen, M. Styblo, D. J. Thomas and A. D. Kligerman, “Methylated Trivalent Arsenic Species are Genotoxic,” Chemical Research Toxicology, vol. 14, no. 4, April 2001, pp. 355- 361.
- U. K. Chowdhury, B. K. Biswas, T. R. Chowdhury, G. Samanta, B. K. Mandal, G. C. Basu, C. R. Chanda, D. Lodh, K. C. Saha, S. K. Mukherjee, et al., “Groundwater Arsenic Contamination in Bangladesh and West Bengal, India,” Environmental Health Perspectives, vol. 108, no. 5, May 2000, pp. 393-397.
- IARC Monographs on the Evaluation of the Carcinogenic Risk of Chemicals to Man-Overall Evaluations of Carcinogenicity: An Update of IARC Monograph 1 to 42: Arsenic and Arsenic Compound, IARC, Lyon, 1987.
- K. T. Kitchin, “Recent Advances in Arsenic Carcinogenesis: Modes of Action, Animal Model Systems, and Methylated Arsenic Metabolites,” Toxicology and Applied Pharmacology, vol. 172, no. 3, 1 May 2001, pp. 249- 261.
- P. B. Tchounwou, A. K. Patlolla and J. A. Centeno, “Carcinogenic and Systemic Health Effects Associated with Arsenic Exposure-A Critical Review,” Toxicologic Pathology, vol. 31, no. 6, November-December 2003, pp. 575-588.
- T. G. Rossman, A. N. Uddin and F. J. Burns, “Evidence that Arsenite Acts as a Cocarcinogen in Skin Cancer,” Toxicology and Applied Pharmacology, vol. 198, no. 3, 1 August 2004, pp. 394-404.
- M. P. Waalkes, J. Liu, D. R. Germolec, C. S. Trempus, R. E. Cannon, E. J. Tokar, R. W. Tennant, J. M. Ward and B. A. Diwan, “Arsenic Exposure in Utero Exacerbates Skin Cancer Response in Adulthood with Contemporaneous Distortion of Tumor Stem Cell Dynamics,” Cancer Research, vol. 68, no. 20, 15 October 2008, pp. 8278- 8285.
- M. Vahter, “Health Effects of Early Life Exposure to Arsenic,” Basic Clinical Pharmacology and Toxicology, vol. 102, no. 2, February 2008, pp. 204-211.
- E. T. Snow, “Metal Carcinogenesis: Mechanistic Implications,” Pharmacology & Therapeutics, vol. 53, no. 1, 1992, pp. 31-65.
- M. Lu, H. Wang, Z. Wang, X. F. Li and X. C. Le, “Identification of Reactive Cysteines in a Protein Using Arsenic Labeling and Collision-Induced Dissociation Tandem Mass Spectrometry,” Journal of Proteome Research, vol. 7, no. 8, August 2008, pp. 3080-3090.
- C. O. Abernathy, Y. P. Liu, D. Longfellow, H. V. Aposhian, B. Beck, B. Fowler, R. Goyer, R. Menzer, T. Rossman, C. Thompson, et al., “Arsenic: Health Effects, Mechanisms of Actions and Research Issues,” Environmental Health Perspectives, vol. 107, no. 7, July 1999, pp. 593-597.
- D. Zhang, J. Li, J. Gao and C. Huang, “c-Jun/AP-1 Pathway-Mediated Cyclin D1 Expression Participates in Low Dose Arsenite-Induced Transformation in Mouse Epidermal JB6 Cl41 Cells,” Toxicology and Applied Pharmacology, vol. 235, no. 1, 15 February 2009, pp. 18-24.
- F. G. Burleson, P. P. Simeonova, D. R. Germolec and M. I. Luster, “Dermatotoxic Chemical Stimulate of c-jun and c-fos Transcription and AP-1 DNA Binding in Human Keratinocytes,” Research Communications in Molecular Pathology & Pharmacology, vol. 93, no. 2, August 1996, pp. 131-148.
- M. Cavigelli, W. W. Li, A. Lin, B. Su, K. Yoshioka and M. Karin, “The Tumor Promoter Arsenite Stimulates AP-1 Activity by Inhibiting a JNK Phosphatase,” EMBO Journal, vol. 15, no. 22, 15 November 1996, pp. 6269- 6279.
- H. S. Yu, K. S. Chiou, G. S. Chen, R. C. Yang and S. F. Chang, “Progressive Alterations of Cytokeratin Expressions in the Process of Chronic Arsenism,” Journal of Dermatology, vol. 20, no. 12, December 1993, pp. 741- 745.
- M. Matsui, C. Nishigori, S. Toyokuni, J. Takada, M. Akaboshi, M. Ishikawa, S. Imamura and Y. Miyachi, “The Role of Oxidative DNA Damage in Human Arsenic Carcinogenesis: Detection of 8-Hydroxy-2’-Deoxyguanosine in Arsenic-Related Bowen’s Disease,” Journal of Investigative Dermatology, vol. 113, no. 1, July 1999, pp. 26-31.
- M. Yamamoto, S. Hirano, C. F. Vogel, X. Cui and F. Matsumura, “Selective Activation of NF-Kappab and E2F By Low Concentration of Arsenite in U937 Human Monocytic Leukemia Cells,” Journal of Biochemical and Molecular Toxicology, vol. 22, no. 2, 2008, pp. 136-146.
- A. Barchowsky, E. J. Dudek, M. D. Treadwell and K. E. Wetterhahn, “Arsenic Induces Oxidant Stress and NF-Kappa B Activation in Cultured Aortic Endothelial Cells,” Free Radical Biology and Medicine, vol. 21, no. 6, 1996, pp. 783-790.
- C. Huang, J. Li, M. Ding, L. Wang, X. Shi, V. Castranova, V. Vallyathan, G. Ju and M.Costa, “ArsenicInducedNfkappab Transactivation Through Erksand JnksDependent Pathways in Mouse Epidermal Jb6 Cells,” Molecular and Cellular Biochemistry, vol. 222, no. 1-2, June 2001, pp. 29-34.
- J. B. Wijeweera, A. J. Gandolfi, A. Parrish and R. C. Lantz, “Sodium Arsenite Enhances AP-1 and NFkappaB DNA Binding and Induces Stress Protein Expression in Precision-Cut Rat Lung Slices,” Toxicology Science, vol. 61, no. 2, June 2001, pp. 283-294.
- W. T. Liao, K. L. Chang, C. L. Yu, G. S. Chen, L. W. Chang and H. S. Yu, “Arsenic Induces Human Keratinocyte Apoptosis by the FAS/FAS Ligand Pathway, Which Correlates with Alterations in Nuclear Factor-Kappa B and Activator Protein-1 Activity,” Journal of Investigative Dermatology, vol. 122, no. 1, January 2004, pp. 125- 129.
- P. Ghosh, M. Banerjee, S. De Chaudhuri, J. K. Das, N. Sarma, A. Basu and A. K. Giri, “Increased Chromosome Aberration Frequencies in the Bowen's Patients Compared to Non-Cancerous Skin Lesions Individuals Exposed to Arsenic,” Mutation Research, vol. 632, no. 1-2, 15 August 2007, pp. 104-110.
- T. C. Lee, N. Tanaka, P. W. Lamb, T. M. Gilmer and J. C. Barrett, “Induction of Gene Amplification by Arsenic,” Science, vol. 241, no. 4861, 1 July 1988, pp. 79-81.
- J. Mahata, A. Basu, S. Ghoshal, J. N. Sarkar, A. K. Roy, G. Poddar, A. K. Nandy, A. Banerjee, K. Ray, A. T. Natarajan, et al., “Chromosomal Aberrations and Sister Chromatid Exchanges in Individuals Exposed to Arsenic through Drinking Water in West Bengal, India,” Mutation Research, vol. 534, no. 1-2, 10 January 2003, pp. 133- 143.
- R. N. Huang, I. C. Ho, L. H. Yih and T. C. Lee, “Sodium Arsenite Induces Chromosome Endoreduplication and Inhibits Protein Phosphatase Activity in Human Fibroblasts,” Environmental and Molecular Mutagenesis, vol. 25, No. 3, 1995, pp. 188-196.
- T. S. Kochhar, W. Howard, S. Hoffman and L. Brammer-Carleton, “Effect of Trivalent and Pentavalent Arsenic in Causing Chromosome Alterations in Cultured Chinese Hamster Ovary (CHO) Cells,” Toxicology Letters, vol. 84, no. 1, January 1996, pp. 37-42.
- K. Yamanaka and S. Okada, “Induction of Lung-Specific DNA Damage by Metabolically Methylated Arsenics via the Production of Free Radicals,” Environmental Health Perspectives, vol. 102, Supplement 3, September 1994, pp. 37- 40.
- L. H. Yih, and T. C. Lee, “Effects of Exposure Protocols on Induction of Kinetochore-Plus and -Minus Micronuclei by Arsenite in Diploid Human Fibroblasts,” Mutation Research, vol. 440, no. 1, 15 March 1999, pp. 75-82.
- A. Basu, J. Mahata, A. K. Roy, J. N. Sarkar, G. Poddar, A. K. Nandy, P. K. Sarkar, P. K. Dutta, A. Banerjee, M. Das, et al., “Enhanced Frequency of Micronuclei in Individuals Exposed to Arsenic through Drinking Water in West Bengal, India,” Mutation Research, vol. 516, no. 1-2, 26 April 2002, pp. 29-40.
- D. Tian, H. Ma, Z. Feng, Y. Xia, X. C. Le, Z. Ni, J. Allen, B. Collins, D. Schreinemachers and J. L. Mumford, “Analyses of Micronuclei in Exfoliated Epithelial Cells from Individuals Chronically Exposed to Arsenic via Drinking Water in Inner Mongolia, China,” Journal of Toxicology and Environmental Health A, vol. 64, no. 6, 23 November 2001, pp. 473-484.
- M. L. Warner, L. E. Moore, M. T. Smith, D. A. Kalman, E. Fanning and A. H. Smith, “Increased Micronuclei in Exfoliated Bladder Cells of Individuals Who Chronically Ingest Arsenic-Contaminated Water in Nevada,” Cancer Epidemiol Biomarkers Prevention, vol. 3, no. 7, OctoberNovember 1994, pp. 583-590.
- C. W. Chien, M. C. Chiang, I. C. Ho and T. C. Lee, “Association of Chromosomal Alterations with ArseniteInduced Tumorigenicity of Human Hacat Keratinocytes in Nude Mice,” Environmental Health Perspectives, vol. 112, no. 17, December 2004, pp. 1704-1710.
- X. J. Qin, L. G. Hudson, W. Liu, W. Ding, K. L. Cooper, and K. J. Liu, “Dual Actions Involved in Arsenite-Induced Oxidative DNA Damage,” Chemical Research Toxicology, vol. 21, no. 9, September 2008, pp. 1806-1813.
- S. F. Lee-Chen, C. T. Yu and K. Y. Jan, “Effect of Arsenite on the DNA Repair of UV-Irradiated Chinese Hamster Ovary Cells,” Mutagenesis, vol. 7, no. 1, January 1992, pp. 51-55.
- H. K. Hamadeh, K. J. Trouba, R. P. Amin, C. A. Afshari and D. Germolec, “Coordination of Altered DNA Repair and Damage Pathways in Arsenite-Exposed Keratinocytes,” Toxicology Science, vol. 69, no. 2, October 2002, pp. 306-316.
- J. H. Li and T. G. Rossman, “Inhibition of DNA Ligase Activity by Arsenite: A Possible Mechanism of its Comutagenesis,” Molecular Toxicology, vol. 2, no. 1, Winter 1989, pp. 1-9.
- Y. Hu, L. Su and E. T. Snow, “Arsenic Toxicity Is Enzyme Specific and its Affects on Ligation are Not Caused by the Direct Inhibition of DNA Repair Enzymes,” Mutation Research, vol. 408, no. 3, 11 September 1998, pp. 203-218.
- S. Lynn, H. T. Lai, J. R. Gurr and K. Y. Jan, “Arsenite retards DNA Break Rejoining by Inhibiting DNA Ligation,” Mutagenesis, vol. 12, no. 5, September 1997, pp. 353-358.
- J. W. Yager and J. K. Wiencke, “Inhibition of Poly (ADP-Ribose) Polymerase by Arsenite,” Mutation Research, vol. 386, no. 3, June 1997, pp. 345-351.
- T. G. Rossman, “Mechanism of Arsenic Carcinogenesis: An Integrated Approach,” Mutation Research, vol. 533, no. 1-2, 10 December 2003, pp. 37-65.
- T. C. Lee, M. Oshimura, and J. C. Barrett, , “Comparison of Arsenic-Induced Cell Transformation, Cytotoxicity, Mutation and Cytogenetic Effects in Syrian Hamster Embryo Cells in Culture,” Carcinogenesis, vol. 6, no. 10, October 1985, pp. 1421-1426.
- T. C. Lee, K. C. Lee, Y. J. Tzeng, R. Y. Huang and K. Y. Jan, “Sodium Arsenite Potentiates the Clastogenicity and Mutagenicity of DNA Crosslinking Agents,” Environmental Mutagenesis, vol. 8, no. 1, 1986, pp. 119-128.
- J. L. Yang, M. F. Chen, C. W. Wu and T. C. Lee, “Posttreatment with Sodium Arsenite Alters the Mutational Spectrum Induced by Ultraviolet Light Irradiation in Chinese Hamster Ovary Cells,” Environmental and Molecular Mutagenesis, vol. 20, no. 3, 1992, pp. 156-164.
- X. Zhou, H. Sun, T. P. Ellen, H. Chen and M. Costa, “Arsenite Alters Global Histone H3 Methylation,” Carcinogenesis, vol. 29, no. 9, September 2008, pp. 1831- 1836.
- H. Chen, S. Li, J. Liu, B. A. Diwan, J. C. Barrett and M. P. Waalkes, “Chronic Inorganic Arsenic Exposure Induces Hepatic Global and Individual Gene Hypomethylation: Implications for Arsenic Hepatocarcinogenesis,” Carcinogenesis, vol. 25, no. 9, September 2004, pp. 1779- 1786.
- S. Chanda, U. B. Dasgupta, D. Guhamazumder, M. Gupta, U. Chaudhuri, S. Lahiri, S. Das, N. Ghosh and D. Chatterjee, “DNA Hypermethylation of Promoter of Gene p53 and p16 in Arsenic-Exposed People with and without Malignancy,” Toxicology Science, vol. 89, no. 2, February 2006, pp. 431-437.
- J. F. Reichard, M. Schnekenburger and A. Puga, “Long term Low-Dose Arsenic Exposure Induces Loss of DNA Methylation,” Biochemical and Biophysical Research Communication, vol. 352, no. 1, 5 January 2007, pp. 188-192.
- Y. Xie, J. Liu, L. Benbrahim-Tallaa, J. M. Ward, D. Logsdon, B. A. Diwan and M. P. Waalkes, “Aberrant DNA Methylation and Gene Expression in Livers of Newborn Mice Transplacentally Exposed to a Hepatocarcinogenic Dose of Inorganic Arsenic,” Toxicology, vol. 236, no. 1-2, 1 July 2007, pp. 7-15.
- J. A. Centeno, F. G. Mullick, L. Martinez, N. P. Page, H. Gibb, D. Longfellow, C. Thompson and E. R. Ladich, “Pathology Related to Chronic Arsenic Exposure,” Environmental Health Perspectives, vol. 110 Supplement 5, October 2002, pp. 883-886.
- S. Yeh, “Skin Cancer in Chronic Arsenicism,” Human Pathology, vol. 4, no. 4, December 1973, pp. 469-485.
- G. Alain, J. Tousignant and E. Rozenfarb, “Chronic Arsenic Toxicity,” International Journal of Dermatology, vol. 32, no. 12, December 1993, pp. 899-901.
- W. P. Tseng, H. M. Chu, S. W. How, J. M. Fong, C. S. Lin and S. Yeh, “Prevalence of Skin Cancer in an Endemic Area of Chronic Arsenicism in Taiwan,” Journal of the National Cancer Institute, vol. 40, no. 3, March 1968, pp. 453-463.
- D. N. Guha Mazumder, R. Haque, N. Ghosh, B. K. De, A. Santra, D. Chakraborty and A. H. Smith, “Arsenic Levels in Drinking Water and the Prevalence of Skin Lesions in West Bengal, India,” International Journal of Epidemiology, vol. 27, no. 5, October 1998, pp. 871-877.
- S. Yeh, S. W. How and C. S. Lin, “Arsenical Cancer of Skin. Histologic Study with Special Reference to Bowen’s Disease,” Cancer, vol. 21, no. 2, Febuary 1968, pp. 312- 339.
- C. H. Lee, C. L. Yu, W. T. Liao, Y. H. Kao, C.Y. Chai, G. S. Chen and H. S. Yu, “Effects and Interactions of Low Doses of Arsenic and UVB on Keratinocyte Apoptosis,” Chemical Research Toxicology, vol. 17, no. 9, September 2004, pp. 1199-1205.
- Y. Miki, T. Kawatsu, K. Matsuda, H. Machino and K. Kubo, “Cutaneous and Pulmonary Cancers Associated with Bowen's Disease,” Journal of the American Academy of Dermatology, vol. 6, no. 1, January 1982, pp. 26-31.
- H. J. Kim, H. G. Min and E. S. Lee, “Bowen’s Diseases and Basal Cell Carcinomas in a Patient,” Journal of Dermatology, vol. 26, no. 10, October 1999, pp. 695-697.
- E. Koh, N. Kondoh, H. Kaihara, H. Fujioka and K. Kitamura, “Ureteral Tumor with Multiple Bowen’s Disease Forty-Two Years after Exposure to Arsenic,” European Urology, vol. 16, no. 5, 1989, pp. 398-400.
- R. C. Yu, K. H. Hsu, C. J. Chen and J. R. Froines, “Arsenic Methylation Capacity and Skin Cancer,” Cancer Epidemiology, Biomarkers & Prevention, vol. 9, no. 11, November 2000, pp. 1259-1262.
- L. Lee and G. Bebb, “A Case of Bowen’s Disease and Small-Cell Lung Carcinoma: Long-Term Consequences of Chronic Arsenic Exposure in Chinese Traditional Medicine,” Environmental Health Perspectives, vol. 113, no. 2, February 2005, pp. 207-210.
- T. Yoshida, H. Yamauchi and G. Fan Sun, “Chronic Health Effects in People Exposed to Arsenic via the Drinking Water: Dose-Response Relationships in Review,” Toxicology and Applied Pharmacology, vol. 198, no. 3, 1 August 2004, pp. 243-252.
- T. T. Kuo, S.Hu, S. K. Lo and H. L. Chan, “p53 Expression and Proliferative Activity in Bowen’s Disease with or without Chronic Arsenic Exposure,” Human Pathology, vol. 28, no. 7, July 1997, pp. 786-790.
- C. H. Chang, R. K. Tsai, G. S. Chen, H. S. Yu and C. Y. Chai, “Expression of bcl-2, p53 and Ki-67 in Arsenical Skin Cancers,” Journal of Cutaneous Pathology, vol. 25, no. 9, October 1998, pp. 457-462.
- E. V. Komissarova and T. G. Rossman, “Arsenite Induced Poly(ADP-Ribosyl)ation of Tumor Suppressor P53 in Human Skin Keratinocytes as a Possible Mechanism for Carcinogenesis Associated with Arsenic Exposure,” Toxicology and Applied Pharmacology, vol. 243, no. 3, 15 March 2010, pp. 399-404.
- L. H. Yih and T. C. Lee, “Arsenite Induces P53 Accumulation through an ATM-Dependent Pathway in Human Fibroblasts,” Cancer Research, vol. 60, no. 22, 15 November 2000, pp. 6346-6352.
- D. Menendez, G. Mora, A. M. Salazar and P. OstroskyWegman, “ATM Status Confers Sensitivity to Arsenic Cytotoxic Effects,” Mutagenesis, vol. 16, no. 5, September 2001, pp. 443-448.
- C. Y. Chai, H. S. Yu, H. T. Yen, K. B. Tsai, G. S. Chen and C. L. Yu, “The Inhibitory Effect of UVB Irradiation on the Expression of P53 and Ki-67 Proteins in Arsenic-Induced Bowen’s Disease,” Journal of Cutaneous Pathology, vol. 24, no. 1, January 1997, pp. 8-13.
- L. L. Hsieh, H. J. Chen, J. T. Hsieh, S. H. Jee, G. S. Chen and C. J. Chen, “Arsenic-related Bowen’s Disease and Paraquat-Related Skin Cancerous Lesions Show No Detectable Ras and P53 Gene Alterations,” Cancer Letter, vol. 86, no. 1, 28 October 1994, pp. 59-65.
- C. H. Hsu, S. A. Yang, J. Y. Wang, H. S. Yu and S. R. Lin, “Mutational Spectrum of P53 Gene in Arsenic-Related Skin Cancers from the Blackfoot Disease Endemic Area of Taiwan,” British Journal of Cancer, vol. 80, no. 7, June 1999, pp. 1080-1086.
- L. H. Yih, S. W. Hsueh, W. S. Luu, T. H. Chiu and T. C. Lee, “Arsenite Induces Prominent Mitotic Arrest via Inhibition of G2 Checkpoint Activation in CGL-2 Cells,” Carcinogenesis, vol. 26, no. 1, January 2005, pp. 53-63.
- L. H. Yih, Y. Y. Tseng, Y. C. Wu and T. C. Lee, “Induction of Centrosome Amplification during Arsenite-Induced Mitotic Arrest in CGL-2 Cells,” Cancer Research, vol. 66, no. 4, 15 February 2006, pp. 2098-2106.
- S. Kawara, M. Takata and K. Takehara, “High Frequency of DNA Aneuploidy Detected by DNA Flow Cytometry in Bowen’s Disease,” Journal of Dermatological Science, vol. 21, no. 1, September 1999, pp. 23-26.
- M. Faris, N. Kokot, K. Latinis, S. Kasibhatla, D. R. Green, G. A. Koretzky and A. Nel, “The c-Jun N-terminal Kinase Cascade Plays a Role in Stress-Induced Apoptosis in Jurkat Cells by Up-Regulating Fas Ligand Expression,” Journal of Immunology, vol. 160, no. 1, 1 January 1998, pp. 134-144.
- A. Zagariya, S. Mungre, R. Lovis, M. Birrer, S. Ness, B. Thimmapaya and R. Pope, “Tumor Necrosis Factor Alpha Gene Regulation: Enhancement of C/Ebpbeta-Induced Activation by c-Jun,” Molecular and Cellular Biology, vol. 18, no. 5, May 1998, pp. 2815-2824.
- A. Kolbus, I. Herr, M. Schreiber, K. M. Debatin, E. F. Wagner and P. Angel, “c-Jun-dependent CD95-L Expression is a Rate-Limiting Step in the Induction of Apoptosis by Alkylating Agents,” Molecular and Cellular Biology, vol. 20, no. 2, January 2000, pp. 575-582.
- A. C. Hermann and C. H. Kim, “Effects of Arsenic on Zebrafish Innate Immune System,” Marine Biotechnology, NY, vol. 7, no. 5, September-October 2005, pp. 494-505.
- D. Ghosh, S. Bhattacharya and S. Mazumder, “Perturbations in the Catfish Immune Responses by Arsenic: Organ and Cell Specific Effects,” Comparative Biochemistry and Physiology-Part C: Toxicology Pharmacology, vol. 143, no. 4, August 2006, pp. 455-463.
- M. Di Gioacchino, N. Verna, L. Di Giampaolo, F. Di Claudio, M. C. Turi, A. Perrone, C. Petrarca, R. MarianiCostantini, E. Sabbioni and P. Boscolo, “Immunotoxicity and Sensitizing Capacity of Metal Compounds Depend on Speciation,” International Journal of Immunopathology and Pharmacology, Vol. 20, No. 2 Supplement 2, AprilJune 2007, pp. 15-22.
- B. Hernandez-Castro, L.M. Doniz-Padilla, M. Salgado Bustamante, D. Rocha, M. D. Ortiz-Perez, M. E. JimenezCapdeville, D. P. Portales-Perez, A. Quintanar-Stephano and R. Gonzalez-Amaro, “Effect of arsenic on regulatory T cells,” Journal of Clinical Immunology, vol. 29, no. 4, July 2009, pp. 461-469.
- T. Yoshida, T.Shimamura and S. Shigeta, “Enhancement of the Immune Response in Vitro by Arsenic,” International Journal of Immunopharmacology, vol. 9, no. 3 1987, pp. 411-415.
- R. Raqib, S. Ahmed, R. Sultana, Y. Wagatsuma, D. Mondal, A. M. Hoque, B. Nermell, M. Yunus, S. Roy, L. A. Persson, et al., “Effects of in Utero Arsenic Exposure on Child Immunity and Morbidity in Rural Bangladesh,” Toxicology Letter, vol. 185, no. 3, 28 March 2009, pp. 197-202.
- C. D. Kozul, K. H. Ely, R. I. Enelow and J. W. Hamilton, , “Low-dose Arsenic Compromises the Immune Response to Influenza a Infection in Vivo,” Environmental Health Perspectives, vol. 117, no. 9, September 2009, pp. 1441- 1447.
- C. D. Kozul, T. H. Hampton, J. C. Davey, J. A. Gosse, A. P. Nomikos, P. L. Eisenhauer, D. J. Weiss, J. E. Thorpe, M. A. Ihnat and J. W. Hamilton, “Chronic Exposure to Arsenic in the Drinking Water Alters the Expression of Immune Response Genes in Mouse Lung,” Environmental Health Perspectives, vol. 117, no. 7, July 2009, pp. 1108-1115.
- H. S. Yu, K. L. Chang, C. L. Yu, C. S. Wu, G. S. Chen and J. C. Ho, “Defective IL-2 Receptor Expression in Lymphocytes of Patients with Arsenic-Induced Bowen's Disease,” Archives Dermatological Research, vol. 290, no. 12, December 1998, pp. 681-687.
- R. Patterson, L. Vega, K. Trouba, C. Bortner and D. Germolec, “Arsenic-induced Alterations in the Contact Hypersensitivity Response in Balb/C Mice,” Toxicology and Applied Pharmacology, vol. 198, no. 3, 1 August 2004, pp. 434-443.
- H. S. Yu, K. L. Chang, C. M. Wang and C. L. Yu, “Alterations of Mitogenic Responses of Mononuclear Cells by Arsenic in Arsenical Skin Cancers,” Journal of Dermatology, vol. 19, no. 11, November 1992, pp. 710-714.
- H. S. Yu, W. T. Liao, K. L. Chang, C. L. Yu and G. S. Chen, “Arsenic Induces Tumor Necrosis Factor Alpha Release and Tumor Necrosis Factor Receptor 1 Signaling in T Helper Cell Apoptosis,” Journal of Investigative Dermatology, vol. 119, no. 4, October 2002, pp. 812-819.
- L. Vega, P. Ostrosky-Wegman, T. I. Fortoul, C. Diaz, V. Madrid and R. Saavedra, “Sodium Arsenite Reduces Proliferation of Human Activated T-Cells by Inhibition of the Secretion of Interleukin-2,” Immunopharmacology and Immunotoxicology, vol. 21, no. 2, May 1999, pp. 203- 220.
- R. Biswas, P. Ghosh, N. Banerjee, J. K. Das, T. Sau, A. Banerjee, S. Roy, S. Ganguly, M. Chatterjee, A. Mukherjee, et al., “Analysis of T-cell Proliferation and Cytokine Secretion in the Individuals Exposed to Arsenic,” Human & Experimental Toxicology, vol. 27, no. 5, May 2008, pp. 381-386.
- G. A. Soto-Pena, A. L. Luna, L. Acosta-Saavedra, P. Conde, L. Lopez-Carrillo, M. E. Cebrian, M. Bastida, E. S. Calderon-Aranda and L. Vega, “Assessment of Lymphocyte Subpopulations and Cytokine Secretion in Children Exposed to Arsenic,” Journal of the Federation of American Societies for Experimental Biology, vol. 20, no. 6, April 2006, pp. 779-781.
- S. W. Burchiel, L. A. Mitchell, F. T. Lauer, X. Sun, J. D. McDonald, L. G. Hudson and K. J. Liu, “Immunotoxicity and Biodistribution Analysis of Arsenic Trioxide in C57Bl/6 Mice Following a 2-week Inhalation Exposure,” Toxicology and Applied Pharmacology, vol. 241, no. 3, 15 December 2009, pp. 253-259.
- A. Lemarie, C. Morzadec, E. Bourdonnay, O. Fardel and L. Vernhet, “Human Macrophages Constitute Targets for Immunotoxic Inorganic Arsenic,” Journal of Immunology, vol. 177, no. 5, 1 September 2006, pp. 3019-3027.
- N. Banerjee, S. Banerjee, R. Sen, A. Bandyopadhyay, N. Sarma, P. Majumder, J. K. Das, M. Chatterjee, S. N. Kabir and A. K. Giri, “Chronic Arsenic Exposure Impairs Macrophage Functions in the Exposed Individuals,” Journal of Clinical Immunology, vol. 29, no. 5, September 2009, pp. 582-594.
- A. L. Luna, L. C. Acosta-Saavedra, L. Lopez-Carrillo, P. Conde, E. Vera, A. De Vizcaya-Ruiz, M. Bastida, M. E. Cebrian and E. S. Calderon-Aranda, “Arsenic Alters Monocyte Superoxide Anion and Nitric Oxide Production in Environmentally Exposed Children,” Toxicology and Applied Pharmacology, 11 March 2010.
- F. Antoine, J. Ennaciri and D. Girard, “Syk is a Novel Target of Arsenic Trioxide (ATO) and is Involved in the Toxic Effect of ATO in Human Neutrophils,” Toxicology in Vitro, vol. 24, no. 3, April 2010, pp. 936-941.
- S. Gupta, L. Yel, D. Kim, C. Kim, S. Chiplunkar and S. Gollapudi, “Arsenic Trioxide Induces Apoptosis in Peripheral Blood T Lymphocyte Subsets by Inducing Oxidative Stress: A Role of Bcl-2,” Molecular Cancer Therapeutics, vol. 2, no. 8, August 2003, pp. 711-719.
- M. E. Gonsebatt, L. Vega, L. A.Herrera, R. Montero, E. Rojas, M. E. Cebrian and P. Ostrosky-Wegman, “Inorganic Arsenic Effects on Human Lymphocyte Stimulation and Proliferation,” Mutation Research, vol. 283, no. 2, October 1992, pp. 91-95.
- D. A. Eastmond and D. Pinkel, “Detection of Aneuploidy and Aneuploidy-Inducing Agents in Human Lymphocytes Using Fluorescence in Situ Hybridization with Chromosome-Specific DNA Probes,” Mutation Research, vol. 234, no. 5, October 1990, pp. 303-318.
- L. Vega, M. E. Gonsebatt and P. Ostrosky-Wegman, “Aneugenic Effect of Sodium Arsenite on Human Lymphocytes in Vitro: An Individual Susceptibility Effect Detected,” Mutation Research, vol. 334, no. 3, June 1995, pp. 365-373.
- Y. H. Hsu, S. Y. Li, H. Y. Chiou, P. M. Yeh, J. C. Liou, Y. M. Hsueh, S. H. Chang and C. J. Chen, “Spontaneous and Induced Sister Chromatid Exchanges and Delayed Cell Proliferation in Peripheral Lymphocytes of Bowen’s Disease Patients and Matched Controls of Arseniasis-Hyperendemic Villages in Taiwan,” Mutation Research, vol. 386, no. 3, June 1997, pp. 241-251.
- W. N. Wen, T. L. Lieu, H. J. Chang, S. W. Wuu, M. L. Yau and K. Y. Jan, “Baseline and Sodium Arsenite-Induced Sister Chromatid Exchanges in Cultured Lymphocytes from Patients with Blackfoot Disease and Healthy Persons,” Human Genetics, vol. 59, no. 3, 1981, pp. 201-203.
- D. Lerda, “Sister-Chromatid Exchange (SCE) among Individuals Chronically Exposed to Arsenic in Drinking Water,” Mutation Research, vol. 312, no. 2, April 1994, pp. 111-120.
- G. A. Soto-Pena and L. Vega, “Arsenic Interferes with the Signaling Transduction Pathway of T Cell Receptor Activation by Increasing Basal and Induced Phosphorylation of Lck and Fyn in Spleen Cells,” Toxicology and Applied Pharmacology, vol. 230, no. 2, 15 July 2008, pp. 216- 226.
- A. D. Kligerman, C. L. Doerr and A. H. Tennant, “Oxidation and Methylation Status Determine the Effects of Arsenic on the Mitotic Apparatus,” Molecular and Cellular Biochemistry, vol. 279, no. 1-2, November 2005, pp. 113-121.
- T. Ramirez, V. Garcia-Montalvo, C. Wise, R. Cea-Olivares, L. A. Poirier and L. A. Herrera, “S-adenosyl-L-methionine is Able to Reverse Micronucleus Formation Induced by Sodium Arsenite and Other Cytoskeleton Disrupting Agents in Cultured Human Cells,” Mutation Research, vol. 528, no. 1-2, 25 July 2003, pp. 61-74.
- G. Tabellini, P. L. Tazzari, R. Bortul, C. Evangelisti, A. M. Billi, T. Grafone, G. Martinelli, M. Baccarani and A. M. Martelli, “Phosphoinositide 3-kinase/Akt Inhibition Increases Arsenic Trioxide-Induced Apoptosis of Acute Promyelocytic and T-cell Leukaemias,” British Journal of Haematology, vol. 130, no. 5, September 2005, pp. 716- 725.
- Y. Zheng, H. Yamaguchi, C. Tian, M. W. Lee, H. Tang, H. G. Wang and Q. Chen, “Arsenic Trioxide (As2O3) Induces Apoptosis through Activation of Bax in Hematopoietic Cells,” Oncogene, vol. 24, no. 20, 5 May 2005, pp. 3339-3347.
- M. E. El-Sabban, R. Nasr, G. Dbaibo, O. Hermine, N. Abboushi, F. Quignon, J. C. Ameisen, F. Bex, H. de The and A. Bazarbachi, “Arsenic-interferon-alpha-triggered Apoptosis in HTLV-I Transformed Cells is Associated with Tax Down-Regulation and Reversal of NF-kappa B Activation,” Blood, vol. 96, no. 8, 15 October 2000, pp. 2849-2855.
- K. Ishitsuka, S. Hanada, K. Uozumi, A. Utsunomiya and T. Arima, “Arsenic Trioxide and the Growth of Human T-cell Leukemia Virus Type I Infected T-cell Lines,” Leukemia and Lymphoma, vol. 37, no. 5-6, May 2000, pp. 649-655.
- D. R. Germolec, J. Spalding, H. S. Yu, G. S. Chen, P. P. Simeonova, M. C. Humble, A. Bruccoleri, G. A. Boorman, J. F. Foley, T. Yoshida, et al., “Arsenic Enhancement of Skin Neoplasia by Chronic Stimulation of Growth Factors,” American Journal of Pathology, vol. 153, no. 6, December 1998, pp. 1775-1785.
- D. R. Germolec, T. Yoshida, K. Gaido, J. L. Wilmer, P. P. Simeonova, F. Kayama, F. Burleson, W. Dong, R. W. Lange and M. I. Luster, “Arsenic Induces Overexpression of Growth Factorsin Human Keratinocytes,” Toxicology and Applied Pharmacology, vol. 141, no. 1, November 1996, pp. 308-318.
- H. T. Yen, L. C. Chiang, K. H. Wen, S. F. Chang, C. C. Tsai, C. L. Yu and H. S. Yu, “Arsenic Induces Interleukin-8 Expression in Cultured Keratinocytes,” Archives Dermatological Research, vol. 288, no. 11, October 1996, pp. 716-717.
- L.Vega, , M. Styblo, R. Patterson, W. Cullen, C. Wang and D. Germolec, “Differential Effects of Trivalent and Pentavalent Arsenicals on Cell Proliferation and Cytokine Secretion in Normal Human Epidermal Keratinocytes,” Toxicology and Applied Pharmacology, vol. 172, no. 3, 1 May 2001, pp. 225-232.
- W. T. Liao, C. L. Yu, E. L. Cheng-Che, C. H. Lee, C. S. Chang, L. W. Chang, H. L. You and H. S. Yu, “Differential Effects of Arsenic on Cutaneous and Systemic Immunity: Focusing on CD4+ Cell Apoptosis in Patients with Arsenic-Induced Bowen’s Disease,” Carcinogenesis, 17 April 2009.
- T. S. Griffith, T. Brunner, S. M. Fletcher, D. R. Green and T. A. Ferguson, “Fas Ligand-Induced Apoptosis as a Mechanism of Immune Privilege,” Science, vol. 270, no. 5239, 17 November 1995, pp. 1189-1192.
- G. F. Murphy, P. A. Krusinski, L. A. Myzak and W. B. Ershler, “Local Immune Response in Basal Cell Carcinoma: Characterization by Transmission Electron Microscopy and Monoclonal Anti-T6 Antibody,” Journal of the American Academy of Dermatology, vol. 8, no. 4, April 1983, pp. 477-485.
- G. M. Woods, R. C. Malley and H. K. Muller, “The Skin Immune System and the Challenge of Tumour Immunosurveillance,” European Journal of Dermatology, vol. 15, no. 2, March-April 2005, pp. 63-69.