Natural Science
Vol.4 No.11(2012), Article ID:24594,9 pages DOI:10.4236/ns.2012.411112
Phylogenetic analysis of the five internal genes and evolutionary pathways of the Greek H3N8 equine influenza virus*
![]()
1Department of Anatomy and Physiology of Farm Animals, Faculty of Animal Science and Aquaculture, Agricultural University of Athens, Athens, Greece; #Corresponding Author: mbountouri@yahoo.gr
2Pfizer Animal Health, Biologicals Development, Sandwich, UK
Received 4 September 2012; revised 27 October 2012; accepted 10 November 2012
Keywords: Equine Influenza; Internal Genes; Evolution Pathways
ABSTRACT
To amplify the NS, NP, PB1, PB2 and PA internal genes of two equine H3N8 influenza A viruses isolated in Greece in 2003 and 2007 five primer pairs were designed. The derived sequences were analysed from a phylogenetic point of view and compared with the evolutionary patters of the HA and NA proteins. Comparison of nucleotide sequences of the five internal genes of the Greek strains showed high similarity (99.3% - 99.7%) to strains isolated from outbreaks in Europe and Asia during 2002-2008. A total of 11 amino acid substitutions of the surface protein NA and the RNP complex proteins were identified in the Greek strains compared to those of progenitor viruses circulating up to 2003. These substitutions were repeated in Chinese and Mongolian isolates from outbreaks in 2007-2008. Notably NS1 protein did not acquired amino acid substitutions and moreover, a stop codon introduced at position 220 was stably maintained in the Greek strains. Phylogenetic trees of the five internal genes did not show the same separation in clades. Greek strains classified them into the American sublineage (as for the PA) Florida clade II (as for the NP, NS1 and PB1) and among Chinese strains of 2007-2008 outbreaks (as for the PB2). Additionally, evolutionary profiles of these internal proteins, except PB2, indicated a parallel evolution fashion to the HA protein, suggesting the possible occurrence of genetic reassortment between H3N8 viruses of district evolutionary lineages. In conclusion, phylogenetic analysis of the internal genes reported in this study could establish a candidate framework for future scientific communications on the phylogenetic diversity and evolution of the equine influenza viruses.
1. INTRODUCTION
Equine influenza activity caused by H3N8 virus has increased not only in enzootic areas of Europe and the United States, but also in non-enzootic or even naive areas such as Japan and Australia [1,2].
It has been reported that, like human influenza viruses, the haemagglutinin (HA) glycoprotein of equine H3N8 influenza virus underwent gradual antigenic change. In addition to the evolution of HA, the segmented nature of the influenza virus genome allows reassortment to take place resulting in rapid virus evolution [3]. Reassortment between EIVs of different lineages or clades has been reported [4,5] supporting the division of equine influenza viruses into lineages which co-circulate among populations. Moreover, the evolution of the HA gene of equine influenza viruses seems to be more complex than expected as there are reports of equine H3N8 viruses been in a state of “frozen evolution” or been periodically reintroduced into the horse population from an unknown source [6].
Phylogenetic analysis of HA and NA proteins of the Greek isolates suggested reassortment between Eurasian and Florida clade II sublineage [5]. However, the remaining internal genes of these viruses have not been analysed. Since the internal genes are more conserved than the external HA and NA genes, identification of an influenza virus based on the internal genes could be easier than on the external genes.
The functional ribonucleoprotein (RNP) complex of influenza viruses responsible for transcription and replication of viral RNA exists as a heterotrimer polymerase complex consisting of the PB2, PB1 and PA proteins bound to viral RNA and the nucleoprotein (NP). Extensive analysis of protein subunits of the RNP complex of type A viruses has resulted in a fairly detailed understanding of the functional role of each subunit. Briefly, the PB1 protein has been shown to possess RNA polymerase catalytic activity, whereas the PB2 protein is responsible for the binding, cleavage and recruitment of cellular mRNA cap-1 structures essential for viral mRNA synthesis. Although the PA protein has been shown to be essential in influenza virus gene expression, its function has not been fully elucidated. However, experiments have provided evidence indicating that the PA protein is required for switching from mRNA synthesis to cRNA synthesis during virus replication and the ability to induce generalized proteolysis has been demonstrated. The non-structural protein (NS1) has been shown to be a virulence factor in influenza A virus infection in vivo, so it is of particular interest when studying the relative pathogenicity of isolated viruses. It functions as an antagonist of the innate immune response by blocking the activation of interferon via regulatory factor-3 and inhibiting the post-transcriptional processing of cellular mRNAs. NS1 proteins from some influenza viruses are also able to inhibit cellular gene expression, inhibiting the action of IFN [7].
The aim of the present study was to provide a complete profile of protein variability of NP, NS, PB2, PB1 and PA gene segments of the Greek H3N8 equine influenza viruses. In order to achieve our aims five primerpairs were designed and phylogenetic analysis was conducted for their proteins (NP, NS1, PB2, PB1 and PA).
Moreover, the evolution pathways of the internal genes are analysed in relation to the evolutionary pathways of the genes encoding the two surface glycoproteins (HA and NA).
2. MATERIALS & METHODS
2.1. Viruses
Two isolates, A/equine/Athens/03 and A/equine/Athens/07, derived from two outbreaks (2003 and 2007) in the same location in Greece were used in this study. Both viruses were isolated after three passages from RT-PCR and Directigen FluA positive nasal swabs of diseased unvaccinated horses. A/equine/Athens/03 and A/equine/ Athens/07 were grouped together based on their HA and NA protein (Bountouri et al., 2011).
2.2. Nucleotide Sequences
Viral RNAs from both cell cultures and nasal swabs were extracted using the QIAmp viral RNA extraction kit (Qiagen) according to the manufacturer’s instructions.
To amplify the genes of the five internal proteins (PB2, PB1, PA, NP, and NS), five primer-pairs were designed, which are shown in Table 1. RT-PCRs for each gene were carried out with the use of the commercial kit SuperScript One-Step RT-PCR with Platinum Taq (Invitrogen SRL). The cycling conditions of RT-PCRs were as follows: 50˚C for 50 min 94˚C for 2 min, then 40 cycles of 94˚C for 1 min, 55˚C for 1 min (NP, PA genes)/55˚C for 30 sec (PB1, PB2 genes)/54˚C for 1 min (NS gene), 72˚C for 2 min, and a final incubation of 72˚C for 10 min. PCR products were analysed on a 1.5% agarose gel stained with ethidium bromide and purified using the QIAquick PCR Purification Kit (Qiagen) according to the manufacturer’s instructions.
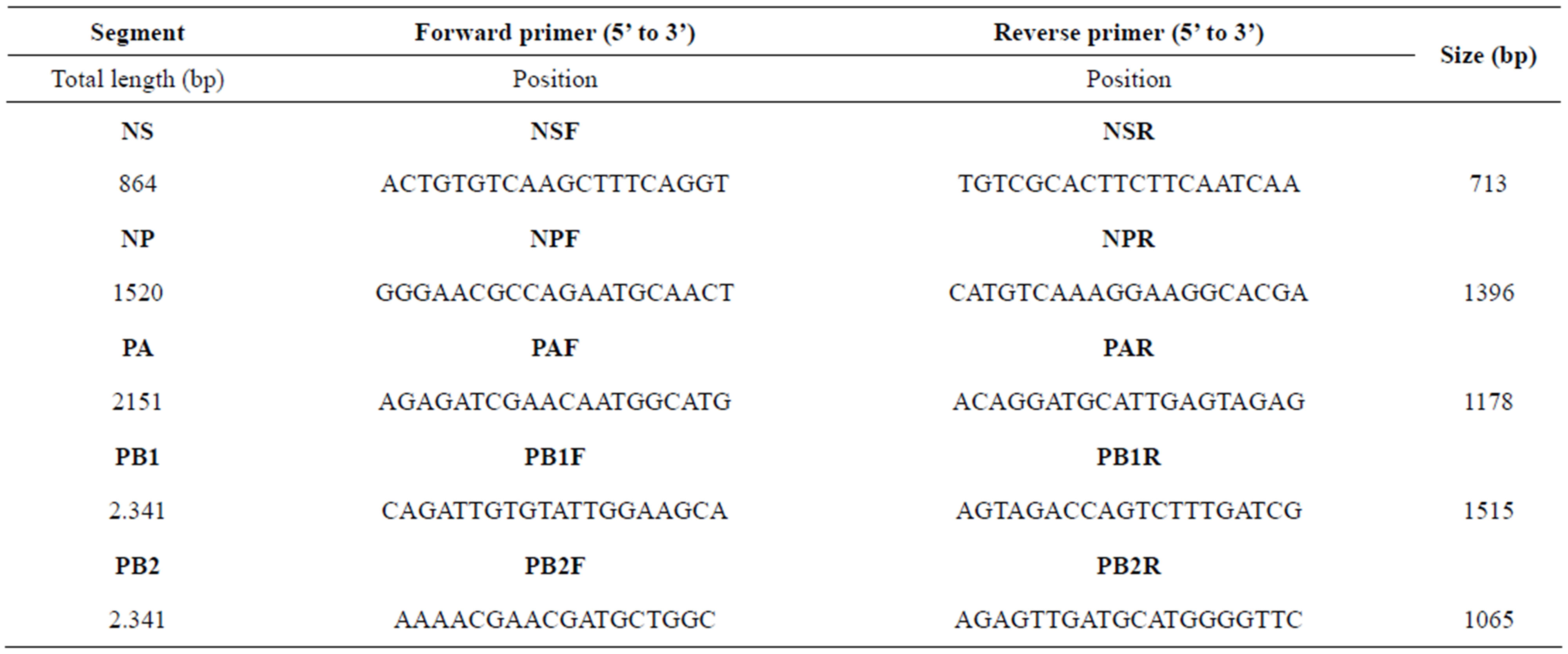
Table 1. Primers designed for this study to amplify gene segments of H3N8 equine influenza virus.
Then they were sequenced by using the PRISM Ready Reaction dye deoxy terminator cycle on an AB3710xl automatic sequencer (Perkin-Elmer Applied Biosystems). Sequences were assembled using Bioedit software package version 3.1 [8] and compared to cognate sequences in the genetic databases using BLAST web-based program (http://www.ncbi.nlm.nih.gov/BLAST). Nucleotides as well as deduced amino acid sequences of all genes were aligned by using Clustal W method.
2.3. Phylogenetic Analysis
All genes examined in the present study were analysed phylogenetically by the neighbour-joining method and by using the MEGA software version 4.0 [9]. To determine the robustness of the trees, the probabilities of the internal branches were estimated by 1000 bootstrap replications.
On the basis of the branching profiles of the phylogenetic trees constructed for the five internal proteins and for the HA and NA proteins of the Greek strains, the distance matrix of the all strains was calculated using Bioedit software and was plotted against the year of the outbreak.
3. RESULTS
3.1. Comparison of Nucleotide and Amino Acid Sequences
The sequences of PB2, PB1, PA, NP and NS genes of the two viruses were determined. The GenBank accession numbers of the sequences reported in this study are HQ696123 and HQ696122 (PB1), HQ696124 and HQ696125 (PB2), HQ696120 and HQ696121 (PA), HQ696116 and HQ696117 (NP), HQ696118 and HQ696119 (NS).
Sequences were compared with published equine influenza virus sequence database using BLAST web-based program. The nucleotide sequences of the five genes of the Greek strains were similar and showed high similarity (99.3% - 99.7%) to those isolated from outbreaks within 2003 and 2007, such as Essex/05 (NS), California/ 02 (NP), Gansu/08 (PA), Xinjiang/07 (PB2) and Newmarket/5/03 (PB1).
In order to identify which amino acids may play a role in separation into different phylogenetic clades, aminoacid sequence alignments of the NS1, NP, PA, PB1 and PB2 coding sequences were constructed with representative isolates aligned against A/eq/Newmarket/5/03. Although the Greek isolates share some common amino acid substitutions with the strain A/Newmarket/5/2003- representative strain of Florida clade II—there were some unique amino acid substitutions which were repeated to isolates from China and Mongolia during outbreaks in 2007 and 2008 (Table 2). Moreover, the previously observed truncation with a stop codon at position 220 of NS1 protein was found in Greek isolates as well.
3.2. Phylogenetic Analysis
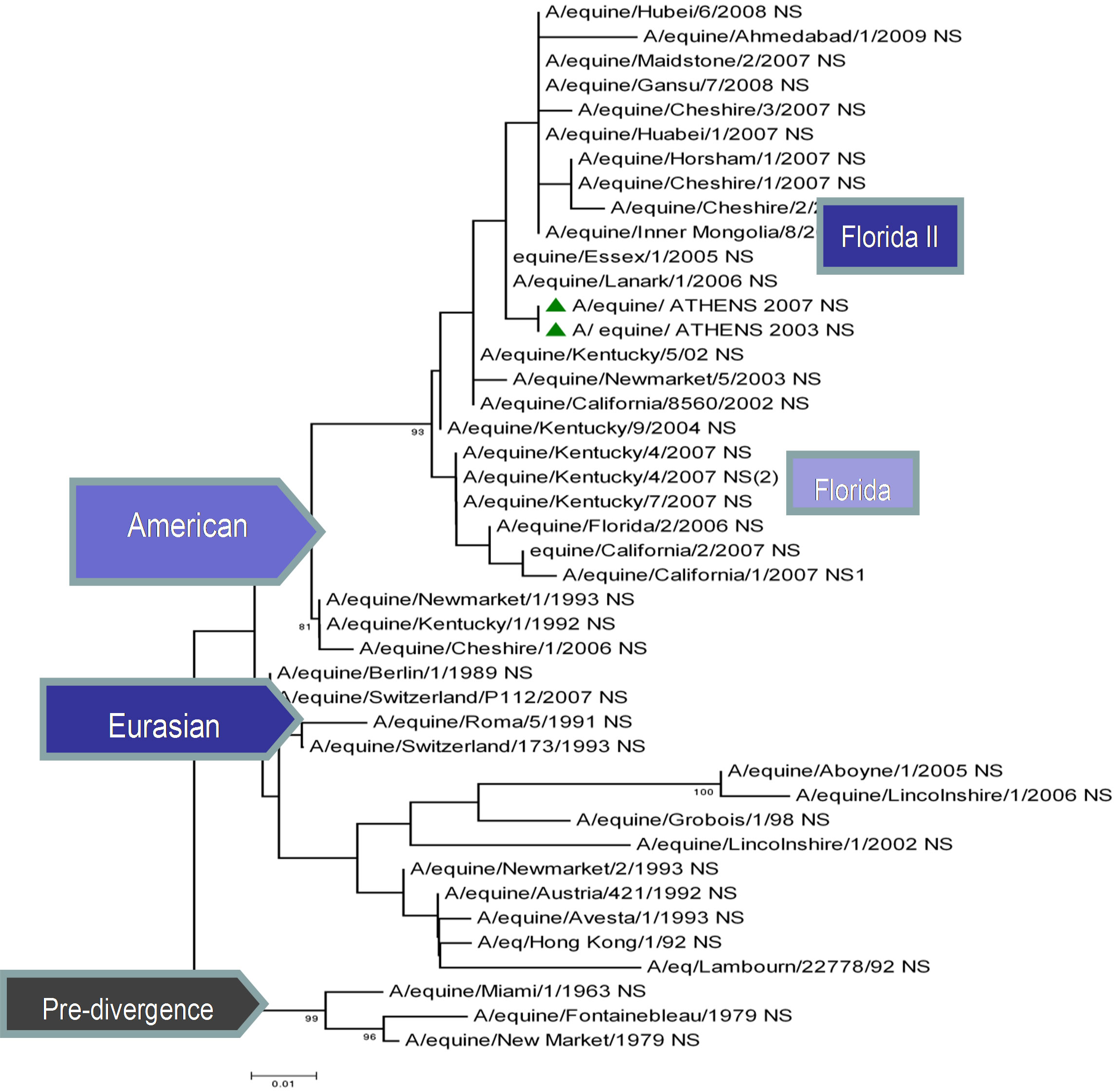
The topology of the maximum-likelihood tree of NS1, ΝΡ and ΡΒ1 proteins showed four clades corresponding to the Pre-divergent, American, Eurasian and Florida lineage viruses similar to those seen with the HA1 coding sequences. There was evidence of further differentiation into two different clades in the Florida sublineage cluster, as seen with HA1, the bootstrap value for this division in NS1 and NP was 100% and in PB1 99% (Figure 1) compared to 100% for HA1 and NA [5]. Whereas the PB2 and PA proteins were less divergent, as PB2 showed no similar classifications onto clades and PA showed three clades, the Pre-divergent, the American and the Eurasian (Figure 1). Thus, the Greek strains were characterised as members of the American lineage and in particular Florida clade II sublineage.
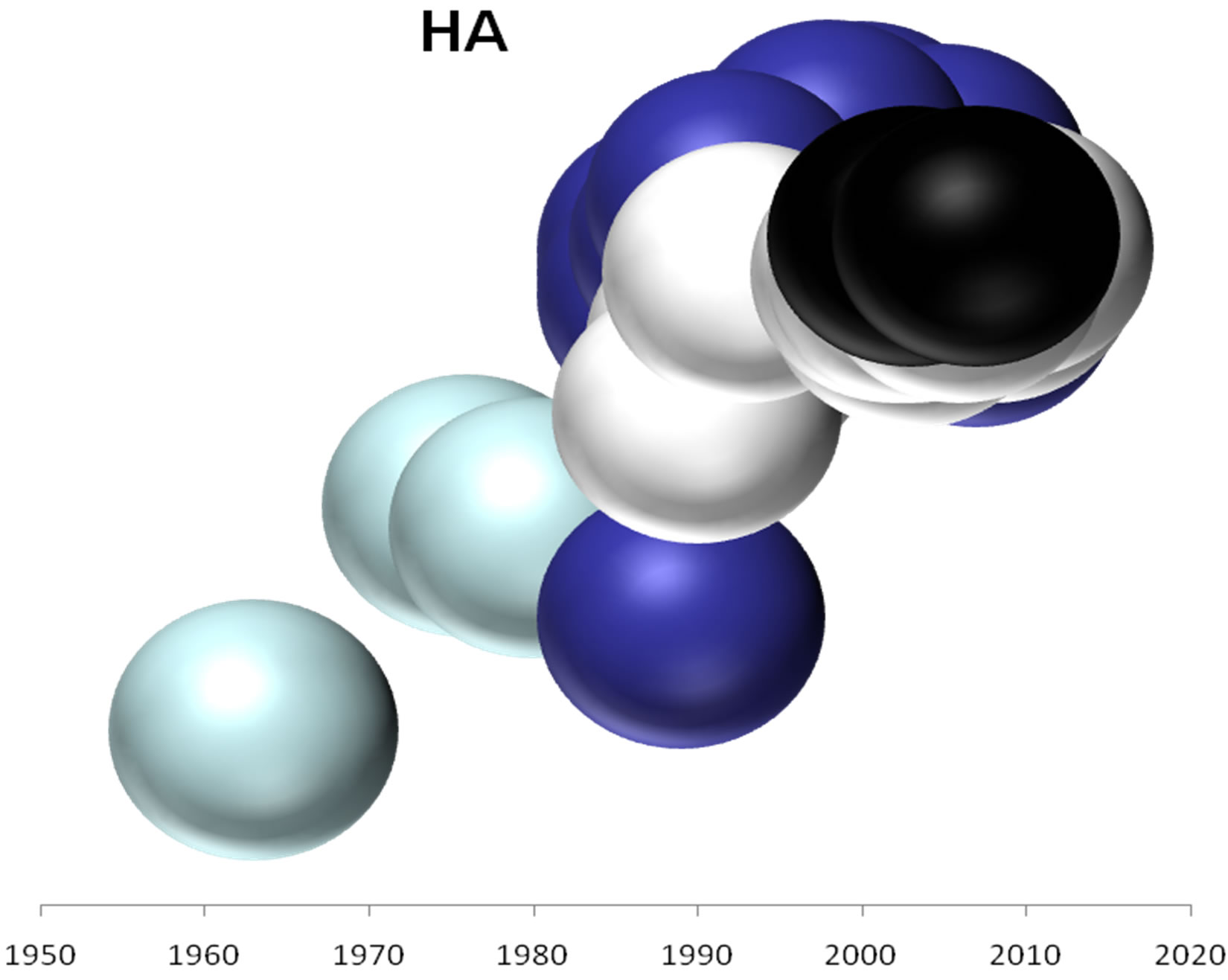
On the basis of the branching profiles of the phylogenetic trees shown in Figure 1, the evolutionary profiles of amino acid sequences of EIVs are illustrated in Figure 2. Additionally, in order to understand the evolutionary
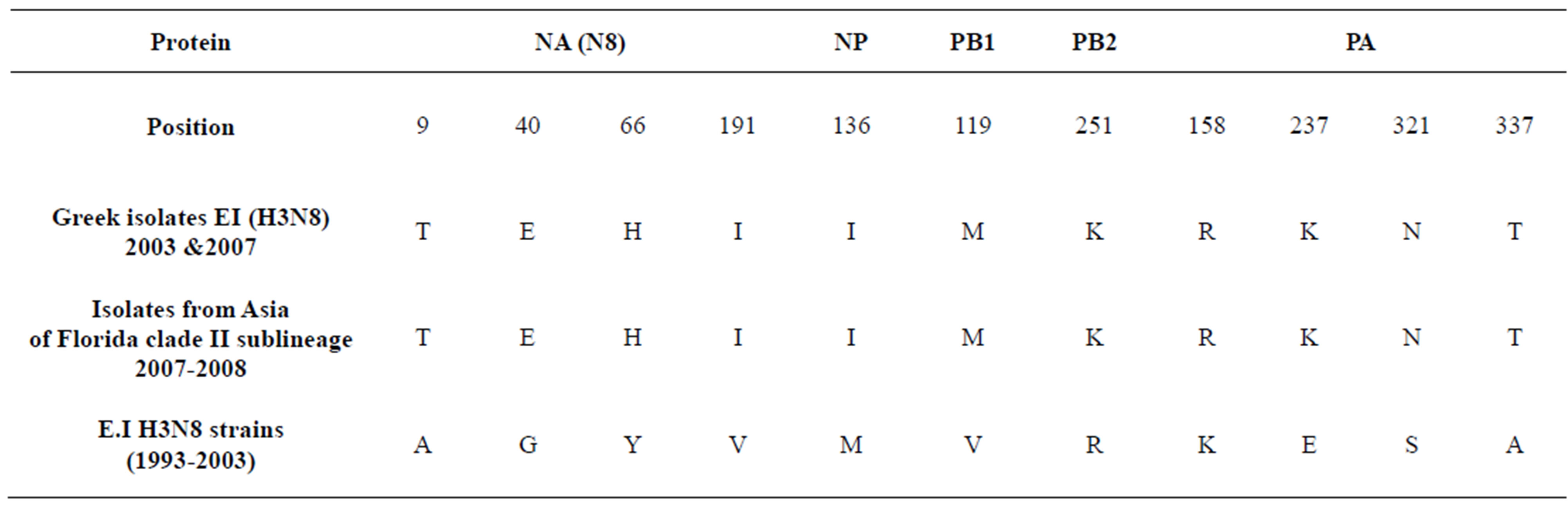
Table 2. Evolution of EIVs.
 (a)
(a)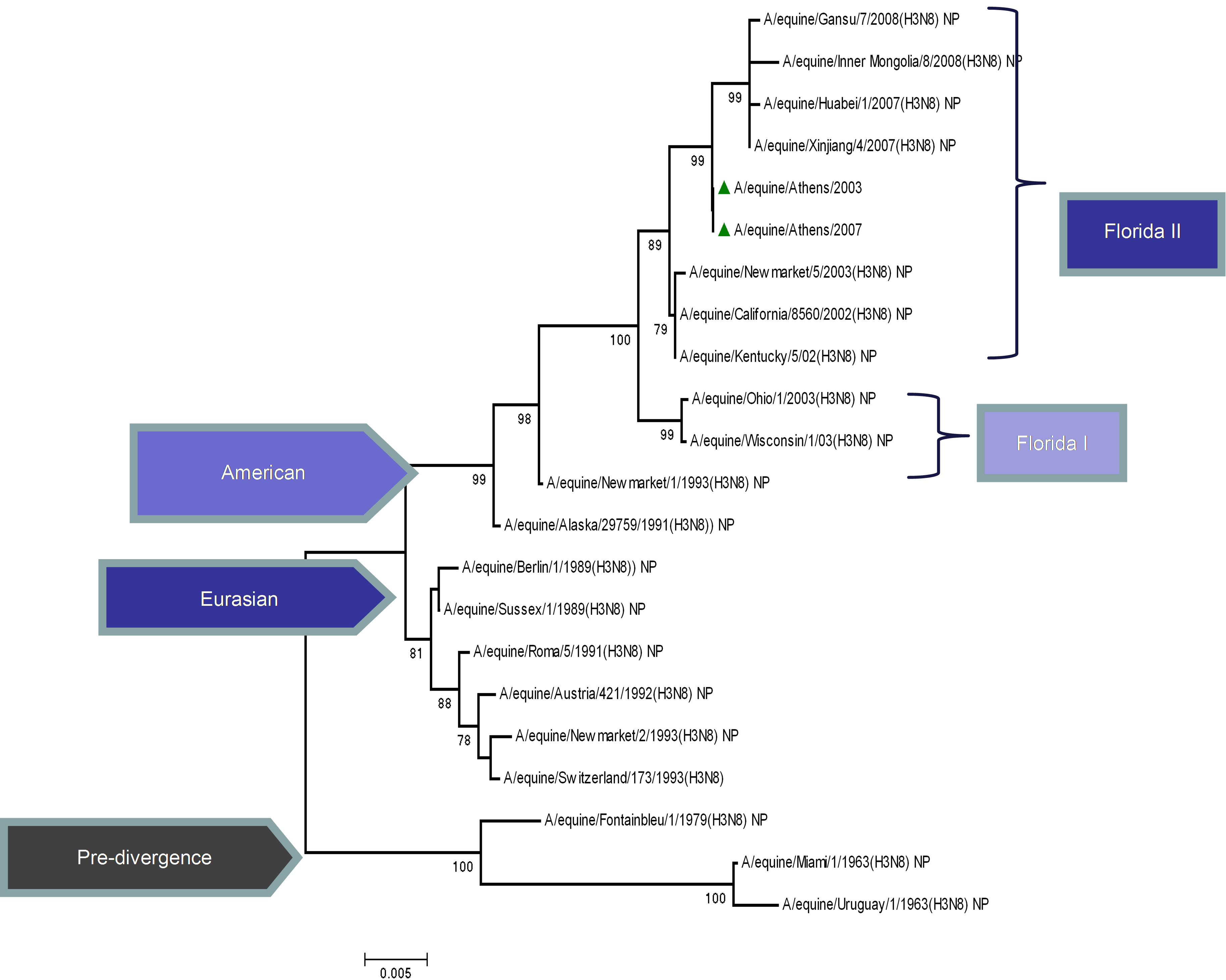 (b)
(b)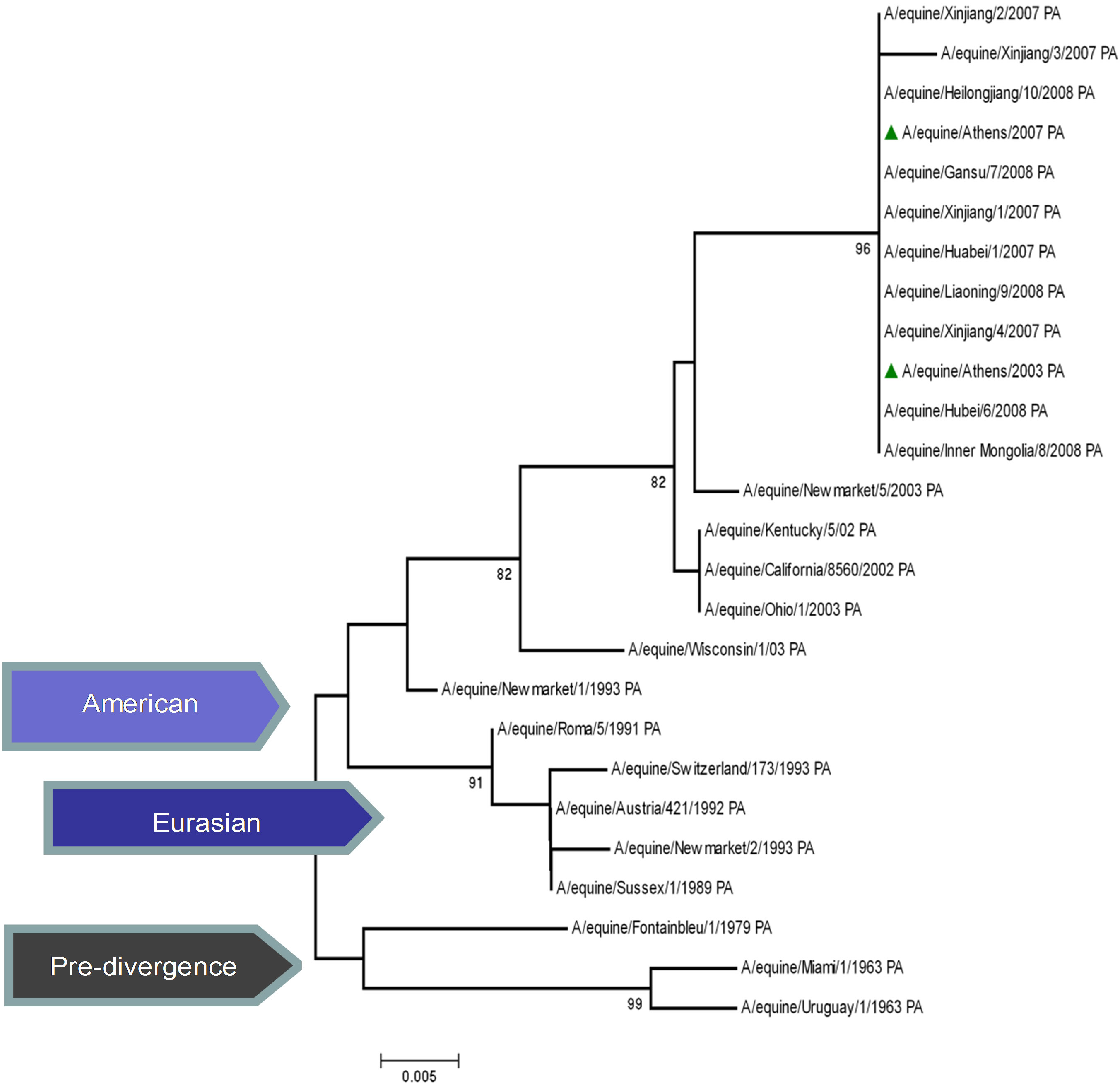 (c)
(c)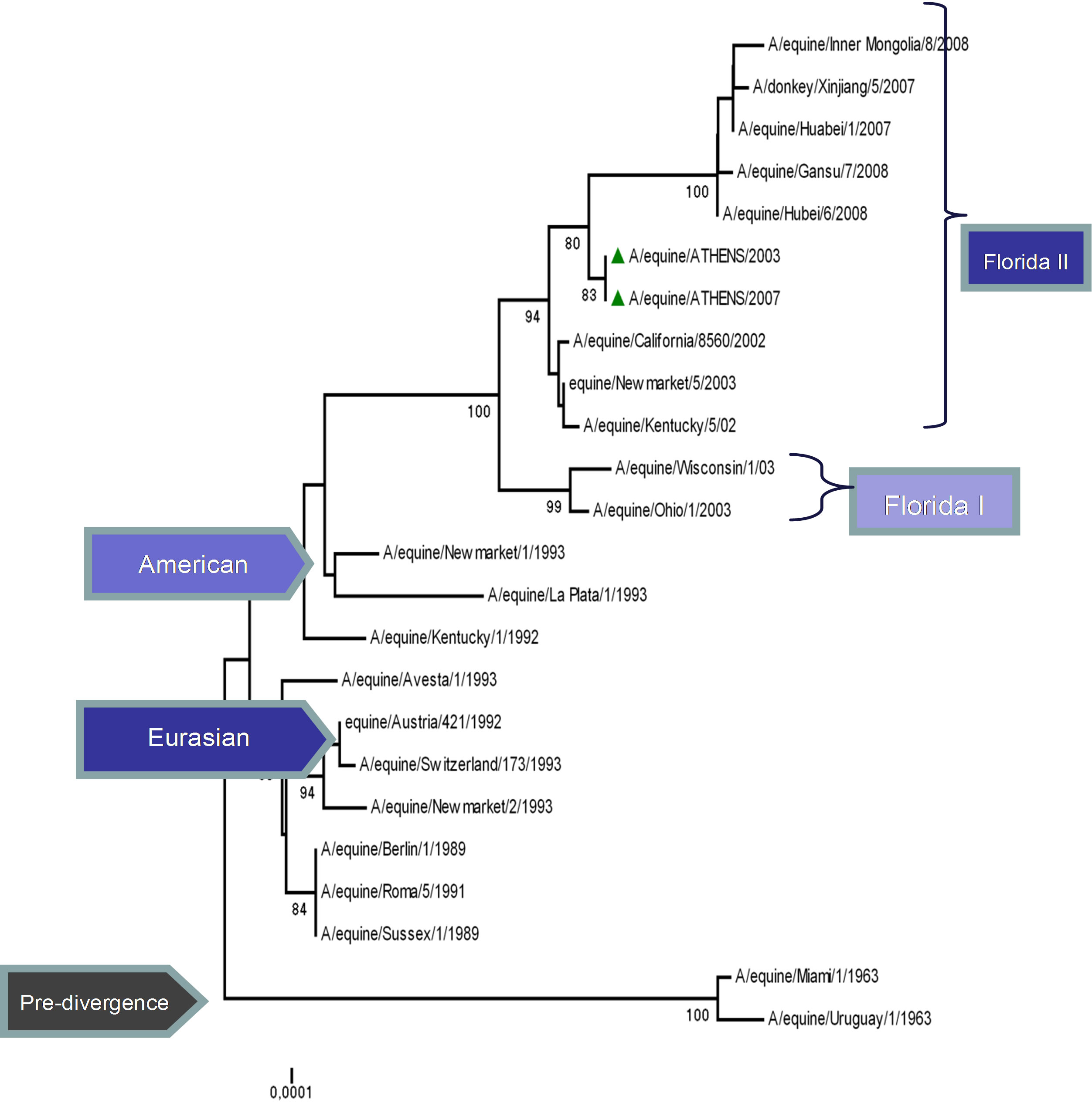 (d)
(d)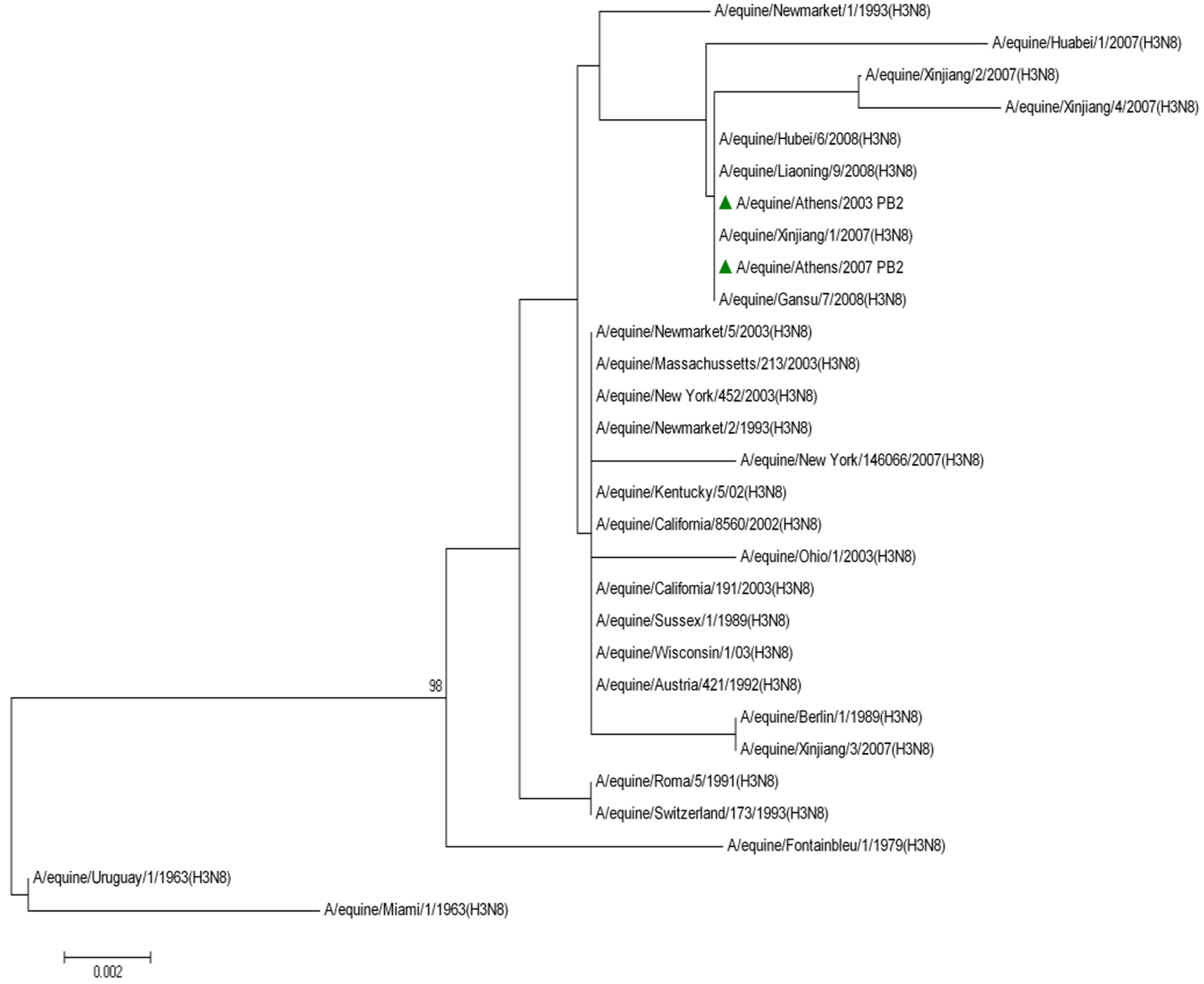 (e)
(e)
Figure 1. Phylogenetic trees of the NS1 (a), NP (b), PA (c), PB1 (d), PB2 (e) proteins of influenza A viruses including those of A/equine/2003 and A/equine/2007 H3N8 viruses. Each tree was constructed by the neighbour-joining method and A/equine/ Miami/1963 was used as the outgroup. Internal branching probabilities were determined by bootstrap analysis using 1000 replications and are indicated as percentages at each branch point.
relationships among the five internal genes of the Greek strains and their HA and NA protein, phylogenetic patterns of these proteins were as well calculated and presented in Figure 2.
4. DISCUSSION
Previous study had shown that reassortment could be responsible for the Eurasian-like HA found in A/equine/ Athens/03 and A/equine/Athens/07, classified as a Florida sublineage clade II virus based on NA amino acid sequence [5]. Thus, in order to trace the origin of the Greek strain, molecular and phylogenetic analysis was conducting for the internal genes of the RNP complex and NS gene. Results revealed that the causative agent was a Florida clade II-like strain which acquired an HA gene from a circulating Eurasian strain.
Molecular analysis of nucleotide and amino acid sequences of the five genes showed that the Greek strain was most similar to equine influenza viruses isolated during outbreaks in Europe and Asia the last decade.
Multiple alignment of Greek isolates with other international revealed eleven amino acid codons that were highly conserved in the previous EIVs H3N8, but were mutated in the Greek strain’s proteins (Table 2). These mutations were as well seen in the strains isolated in China and Mongolia in 2007 and 2008, which belong to the Florida sublineage clade II. Qi et al., report unique aminoacid substitutions of HA protein of Chinese and Mongolian strains isolated in 2007-2008 among Florida clade II strains, suggesting further diversity between the sublineages [10]. Coupled with our findings, it is apparent that amino acid differences occur not only in the surface HA protein, but also in the internal proteins of H3N8 viruses. The NA and PA proteins showed the greatest number of changes. The amino acid substitutions in virion surface protein NA and the polymerase complex subunits are likely to carry functional significance as they are most frequently associated with transmission and virulence [11]. Further study of the virulence and transmission of EI virus in relation to specific genetic changes of internal genes should be undertaken in more
 (a)
(a)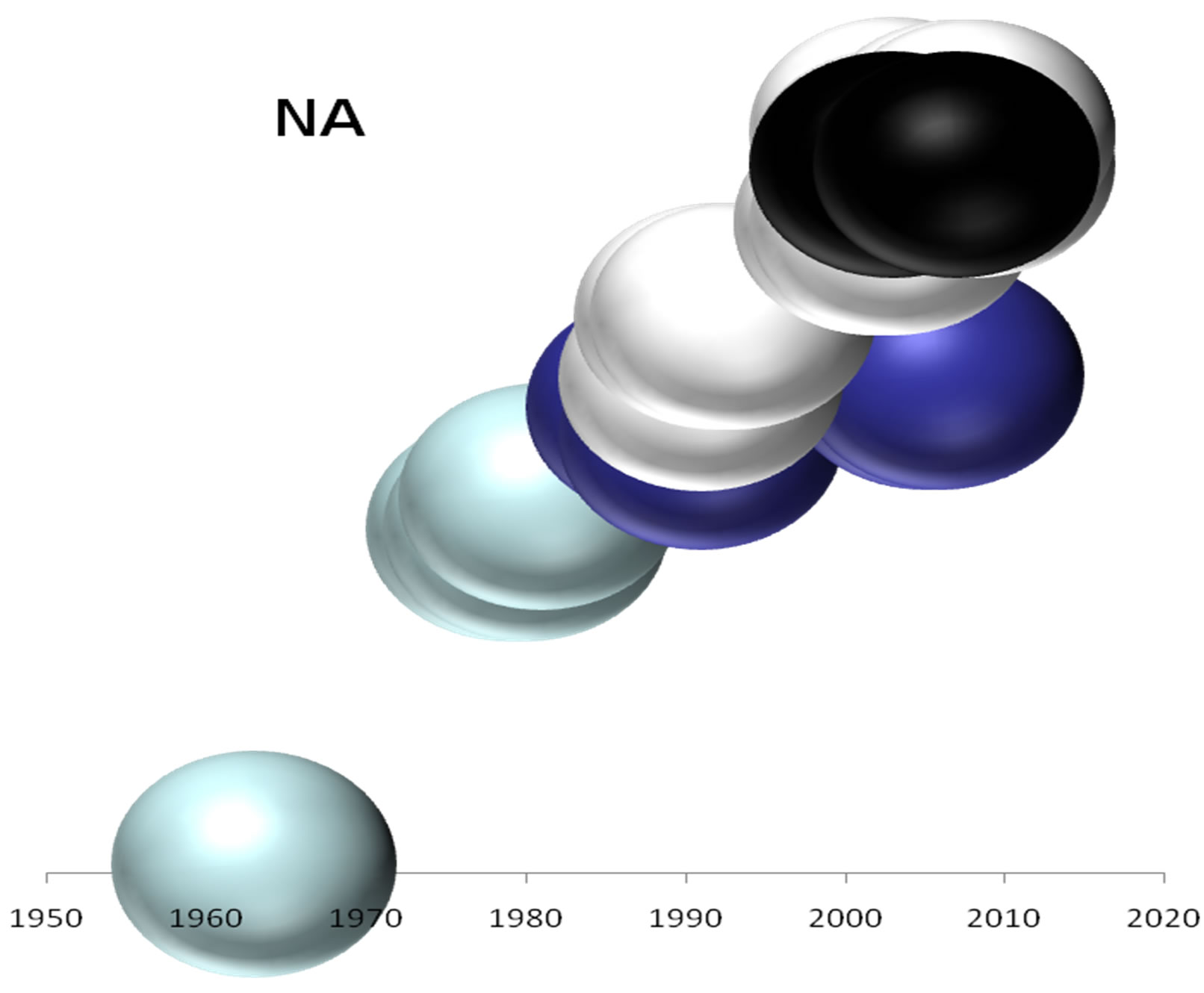 (b)
(b)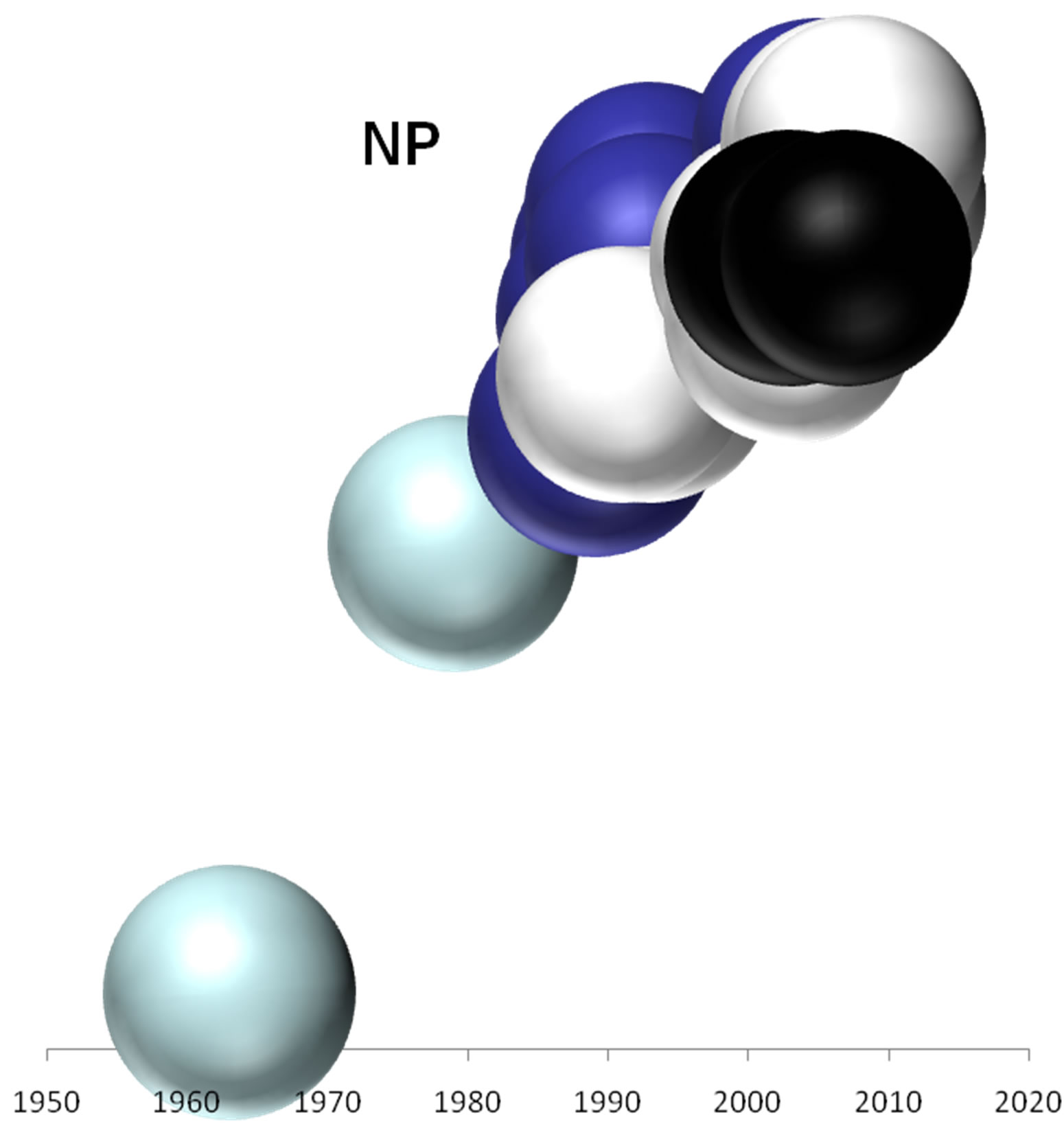 (c)
(c)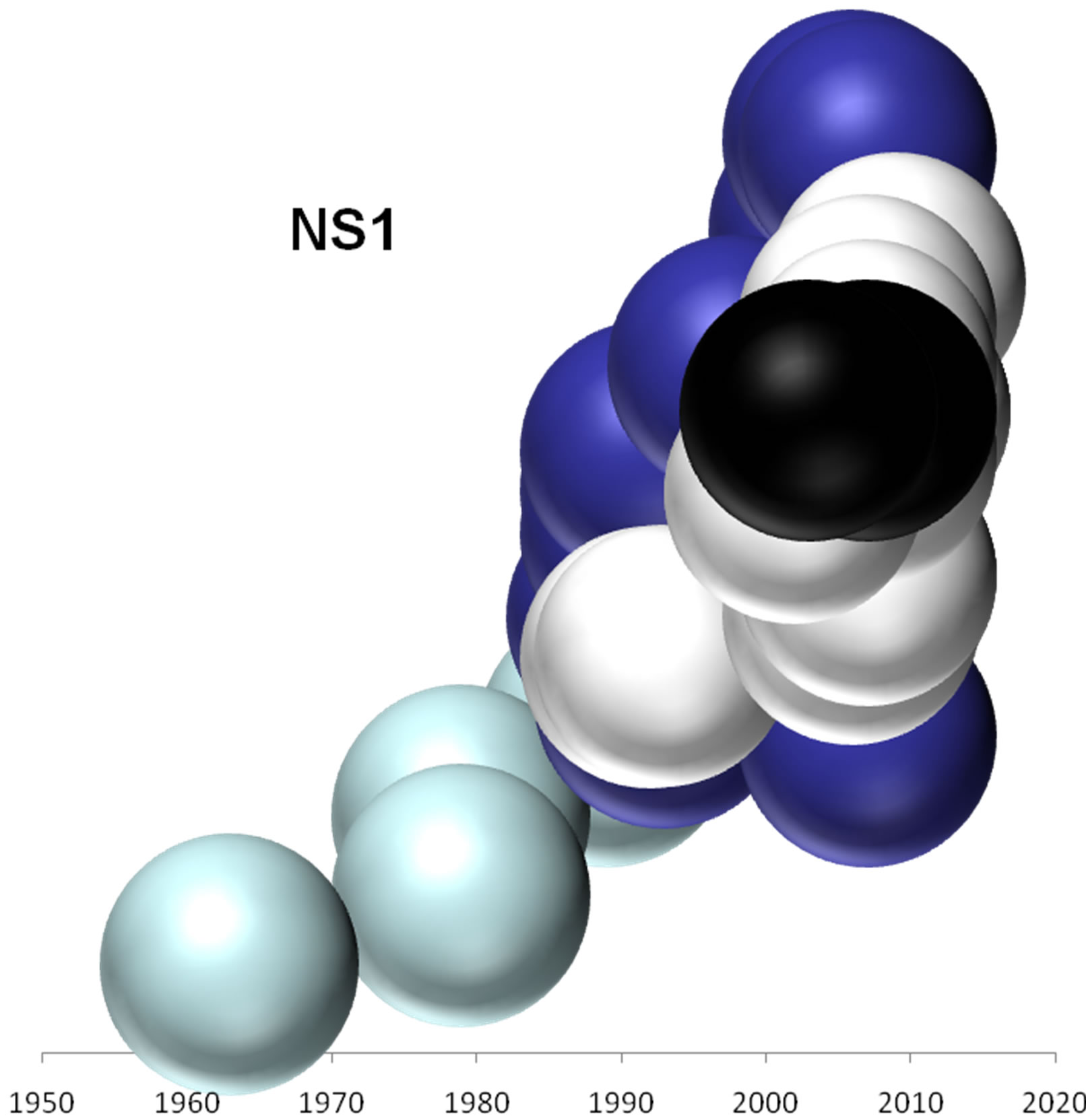 (d)
(d) (e)
(e)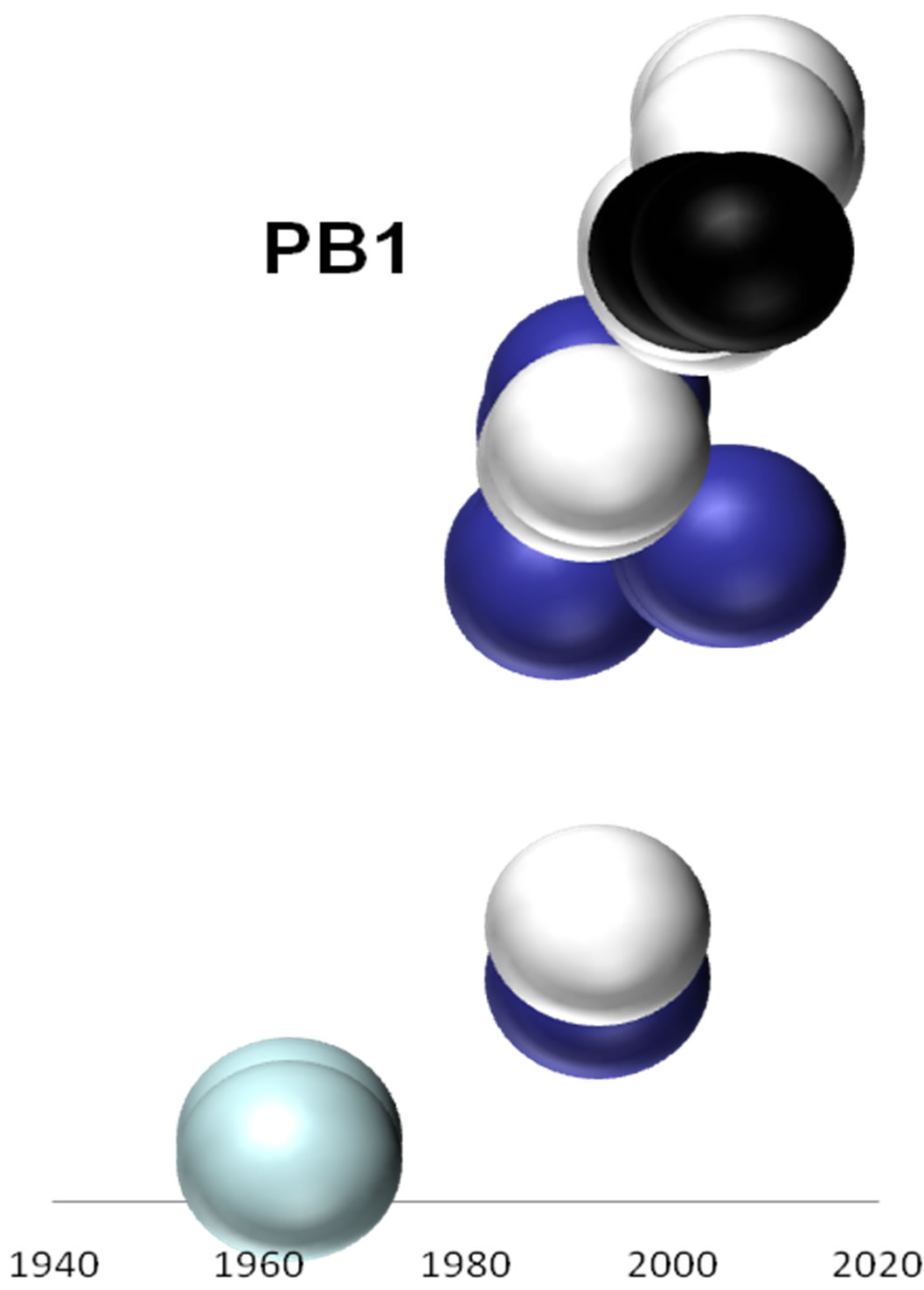 (f)
(f)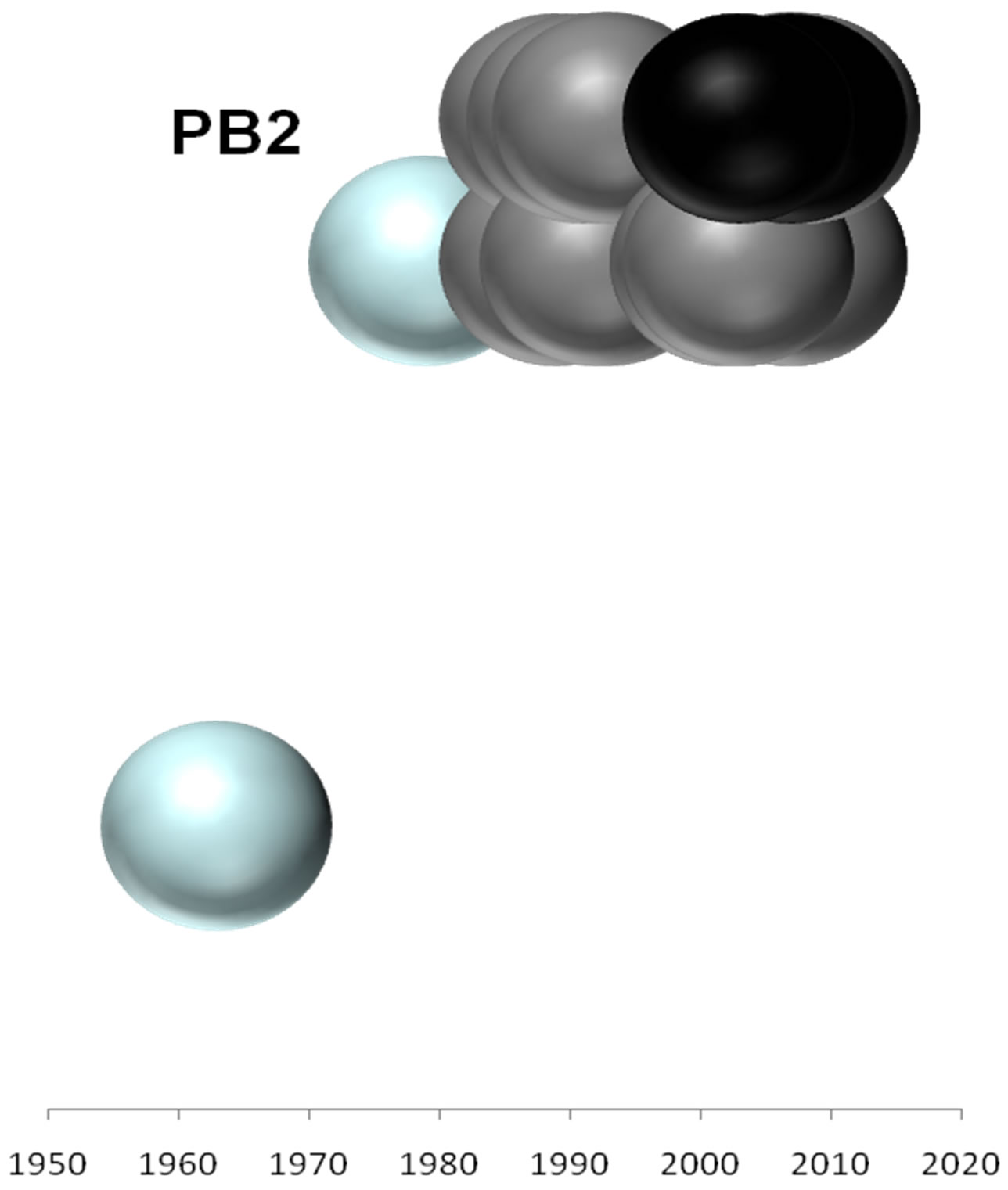 (g)
(g)
Figure 2. Evolutionary profiles for HA (a), NA (b), NS1 (c), NP (d), PB1 (e), PA (f) and PB2 (g) proteins. Based on the plylogenetic tree shown in Figure 1, the distance of each strain from the common ancestor A/equine/ Miami/1963 is plotted against the year of the outbreak. Strains that belong to pre-divergent lineage are shown with light blue colour. Strains that belong to Eurasian lineage are shown with blue colour. Strains that belong to American lineage are shown with white colour. Greek strains are shown with black colour. The strains were characterised members of a lineage according to their HA protein classification.
details. Sequencing of the NS segment showed the truncation, with a premature termination codon at position 220 [4], that was previously observed in EIVs isolated after 2002. This deletion resulted in the loss of a PDZ domain, a widely distributed protein motif that mediates protein-protein interactions in cell signaling and modulates influenza virus replication [12,13].
Phylogenetic analysis classified the Greek strains as members of the Florida sublineage clade II or American lineage according to NS1, NP and PB1 proteins and PA, respectively and among strains isolated in China in 2007 and 2008 according to PB2 protein. Phylogenetic trees of NS1, NP, PB1, PB2 and PA showed the strain A/equine/ Newmarket/5/03 as progenitor of the Greek strains and the last were the immediate ancestors of the Chinese 2007- 2008 strains.
Moreover, the topology of the maximum-likehood tree of NS1, NP and PB1 showed similar classification as has been described for the HA1 and NS1 protein [4]. The three polymerase genes encoding the interacting subunits of the polymerase RNP complex appeared not to be evolving as a unit, supporting previous analysis of the polymerase genes of influenza A virus of various host animals [14,15]. In addition to, the PB2 protein was shown to be less divergent, which correlates with previous evidence [15].
Therefore, the conservatism of the PB2 gene explains the lack of host specificity and may not be an obstacle in adapting the virus to another host species. Nevertheless enzootic canine H3N8 strains isolated the period 2005- 2008 compared with EIV strains showed the greatest changes in the HA protein and PB2 [16]. These substitutions in the protein PB2 considered involved in optimizing the interaction of the viral RNP complex factors to host factors in the nuclear environment [17]. Probably for the same reason the PB2 protein shows conserved among different strains of equine influenza H3N8 from 1990 to today. Evolution profile of all proteins analyzed in this study, with exception of PB2, showed that EI viruses from both American and Euroasian lineages continue to co-circulate, which has reinforced the importance of continued surveillance in the field in order to identify any new emerging threats to equine population.
5. CONCLUSION
In conclusion, we characterised the five internal genes of two equine influenza viruses which caused respiratory disease in Greece. Moreover, both the designed primerpairs and phylogenetic analysis of the internal genes could establish a candidate framework for future scientific communications on the phylogenetic diversity and evolution of the equine influenza viruses.
6. ACKNOWLEDGEMENTS
The Pfizer limited for supporting the above research.
REFERENCES
- Ito, M., Nagai, M., Hayakawa, Y., Komae, H., Murakami, N., Yotsuya, S., Asakura, S., Sakoda, Y. and Kida, H. (2008) Genetic analyses of an H3N8 influenza virus isolate, causative strain of the outbreak of equine influenza at the Kanazawa racecourse in Japan in 2007. Journal of Veterinary Medical Science, 70, 899-906. doi:10.1292/jvms.70.899
- Anon, (2008) Summary of the Australian equine influenza outbreak. Veterinary Record, 163, 378. doi:10.1136/vr.163.13.378
- Webster, R.G., Bean, W.J., Gorman, O.T., Chambers, T.M. and Kawaoka, Y. (1992) Evolution and ecology of influenza A viruses. Microbiological Review, 56, 152-179.
- Bryant, N.A., Rash, A.S., Russell, C.A., Ross, J., Cooke, A., Bowman, S., Shona MacRae, A., Lewis, N.S., Paillot, R., Zanoni, R., Meier, H., Griffiths, L.A., Daly, J.M., Tiwari, A., Chambers, T.M., Newton, J.R. and Elton, D.M. (2009) Antigenic and genetic variations in European and North American equine influenza virus strains (H3N8) isolated from 2006 to 2007. Veterinary Microbiology, 38, 41-52. doi:10.1016/j.vetmic.2009.03.004
- Bountouri, M., Fragkiadaki, E., Ntafis, V., Kanellos, T. and Xylouri, E. (2011) Phylogenetic and molecular characterization of equine H3N8 influenza viruses from Greece (2003 and 2007): Evidence for reassortment between evolutionary lineages. Virology Journal, 8, 350. doi:10.1186/1743-422X-8-350
- Endo, A., Pecoraro, R., Sugita, S. and Nerome, K. (1992) Evolutionary pattern of the H3 haemagglutinin of equine influenza viruses: Multiple evolutionary lineages and frozen replication. Archives of Virology, 123, 73-87. doi:10.1007/BF01317139
- Wright, P. F. and Webster, R.G. (2001) Orthomyxoviruses. In: Fields, B.N. and Knipe, D.M. Eds., Fields Virology, 4th Edition, Lippincott Williams & Wilkins, Philadelphia, 1533-1579.
- Hall, T.A. (1999) BioEdit: A user-friendly biological sequence alignment editor and analysis program for Windows 95/98/NT. Nucleic Acids Symposium Series, 41, 95- 98.
- Tamura, K., Dudley, J., Nei, M. and Kumar, S. (2007) MEGA4: Molecular evolutionary genetics analysis (MEGA) software version 4.0. Molecular Biology and Evolution, 4, 1596-1599. doi:10.1093/molbev/msm092
- Qi, T., Guo, W., Huang, W.-Q., Li, H.-M., Zhao, L.-P., Dai, L.-L., He, N., Hao, X.-F. and Xiang, W.-H. (2010) Genetic evolution of equine influenza viruses isolated in China. Archives of Virology, 155, 1425-1432. doi:10.1007/s00705-010-0724-y
- Neumann, G. and Kawaoka, Y. (2006) Host range restricttion and pathogenicity in the context of influenza pandemic. Emerging Infectious Diseases, 12, 881-886. doi:10.3201/eid1206.051336
- Jackson, D., Hossain, M.J., Hickman, D., Perez, D.R. and Lamb, R.A. (2008) A new influenza virus virulence determinant: The NS1 protein four C-terminal residues modulate pathogenicity. Proceedings of the National Academy of Sciences of the United States of America, 105, 4381- 4386. doi:10.1073/pnas.0800482105
- Jemth, P. and Gianni, S. (2007) PDZ domains: Folding and binding. Biochemistry, 46, 8701-8708. doi:10.1021/bi7008618
- Gorman, O.T., Bean, W., Kawaoka, Y. and Webster, R. (1990) Evolution of the nucleoprotein gene of influenza A virus. Journal of Virology, 64, 1487-1497.
- Gorman, O.T., Donis, R., Kawaoka, Y. and Webster, R. (1990) Evolution of influenza A virus PB2 genes: Implications for evolution of the ribonucleoprotein complex and origin of human influenza A virus. Journal of Virology, 64, 4893-4902.
- Rivailler, P., Perry, I.A., Jang, Y., Davis, C.T., Chen, L.M., Dubovi, E.J. and Donis, R.O. (2010) Evolution of canine and equine influenza (H3N8) viruses co-circulating between 2005 and 2008. Virology, 408, 71-79. doi:10.1016/j.virol.2010.08.022
- Guilligay, D., Tarendeau, F., Resa-Infante, P., Coloma, R., Crepin, T., Sehr, P., Lewis, J., Ruigrok, R.W., Ortin, J., Hart, D.J. and Cusack, S. (2008) The structural basis for cap binding by influenza virus polymerase subunit PB2. Nature Structural & Molecular Biology, 15, 500-506. doi:10.1038/nsmb.1421
NOTES
*The GenBank accession numbers of the sequences reported in this study are HQ696123 and HQ696122 (PB1), HQ696124 and HQ696125 (PB2), HQ696120 and HQ696121 (PA), HQ696116 and HQ696117 (NP), HQ696118 and HQ696119 (NS) and HM164056, HM164057, HM164055, HM164054 (NA).