Journal of Modern Physics
Vol.05 No.17(2014), Article ID:51737,-5 pages
10.4236/jmp.2014.517184
Spin-Dependent Transport in Carbon Nanotubes with Chromium Atoms
Stanislav P. Repetsky1, Oleg V. Tretyak2, Iryna G. Vyshivanaya2, Dmitriy K. Cheshkovskiy1
1Department of Physics, Taras Shevchenko National University of Kyiv, Kyiv, Ukraine
2Institute of High Technologies, Taras Shevchenko National University of Kyiv, Kyiv, Ukraine
Email: srepetsky@univ.kiev.ua
Copyright © 2014 by authors and Scientific Research Publishing Inc.
This work is licensed under the Creative Commons Attribution International License (CC BY).
http://creativecommons.org/licenses/by/4.0/



Received 27 August 2014; revised 25 September 2014; accepted 21 October 2014
ABSTRACT
Method is developed for self-consistent calculation of the energy spectrum of free energy and electrical disordered crystals. Processes of electron scattering on the ionic core potential of different sort, fluctuations of charge, spin density and lattice vibrations are taken into account. Electronic states of the system are described using tight binding multiband model. The nature of the spin-dependent electron transport of carbon nanotubes with chromium atoms adsorbed on the surface is explained. The value of the spin polarization of electron transport is determined by the difference of the partial densities of states of electrons with opposite spin projection at the Fermi level and the difference between the relaxation times of electron states. The value of the spin polarization of the electric current increases with increasing of Cr atoms concentration and magnitude of the external magnetic field.
Keywords:
Spin-Dependent Transport, Carbon Nanotubes with Chromium Atoms, Tight Binding Model, Localized Magnetic Moment, Free Energy, Electrical Conductivity Tensor

1. Introduction
After a relatively recently discovered phenomenon of spin-dependent transport research of electrons spin polarization became actual [1] . Much attention has focused on systems based on carbon doped with transition metals. To clarify the mechanism of spin-dependent transport various theoretical models are intensively investigated. In particular, in [1] theoretically high spin polarization of electrons was predicted in carbon nanotubes, and [2] predicted theoretically almost 100% spin polarization of the electric current in carbon nanotubes with impurities of transition metals Cr, V. One of the possible ways of changing properties of carbon nanotubes in order to apply them in nanoelectronics and spintronics is the application of admixtures of other elements. The presence of impurities can lead to a lowering of the symmetry of the crystal lattice and the degeneracy in the electron energy spectrum, as well as the appearance of additional energy gaps, whose width depends on the type of impurities and their concentrations. However, the effect of impurities on the electronic structure and related properties of carbon nanotubes are investigated insufficiently.
In our work, on the basis of a self-consistent tightbinding model electronic structure, free energy, conductivity and spin-dependent transport of carbon nanotubes with chromium impurity are investigated. Electron scattering on potentials of the ionic cores of different types, and the fluctuations of the spin of the electron density and lattice vibrations are taken into account.
In present literature, the studies of spin-dependent transport in carbon nanotubes are qualitatively described on the basis of ideas about the appearance of a gap in energy spectrum near Fermi level [2] . In our paper, numerical calculations of the spin polarization of electric current in carbon nanotubes are held on the basis of rigorous expressions for conductivity.
2. Tensor of Electrical Conductivity
The method of calculation of the energy spectrum of electrons and phonons, free energy and electrical conductivity of disordered crystal described in [3] -[5] .
Electronic correlations in crystals are described in the multi-zone model of strong coupling. Considered the processes of electron scattering on the potential of ionic skeleton, fluctuations of the charge and spin density and lattice vibrations.
Using the formula Kubo, diagram technique for two-time temperature Green’s function and neglecting the contribution of scattering processes on clusters of three or more sites for static electrical conductivity tensor we can get the expression [3] -[5] :
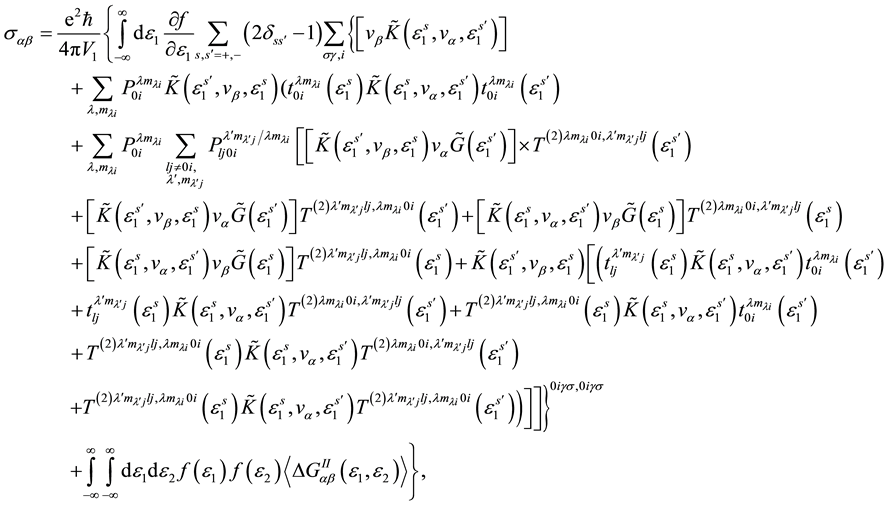 (1)
(1)
where
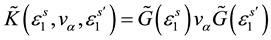 ,
,
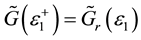
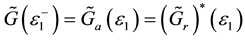 ,
,
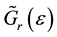 ,
, 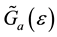 is retarded and advanced one-particle Green functions of effective medium respectively.
is retarded and advanced one-particle Green functions of effective medium respectively.
The first component in the right-hand side of expression (1) describes the contribution of electroconductivity approximation in the coherent potential. Other components describes the contribution in electroconductivity processes of electrons scattering on clusters consisting two atoms.
In the formula (1) component 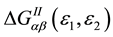 of two-particle Green’s function is caused by the electron-electron interaction and has the form:
of two-particle Green’s function is caused by the electron-electron interaction and has the form:
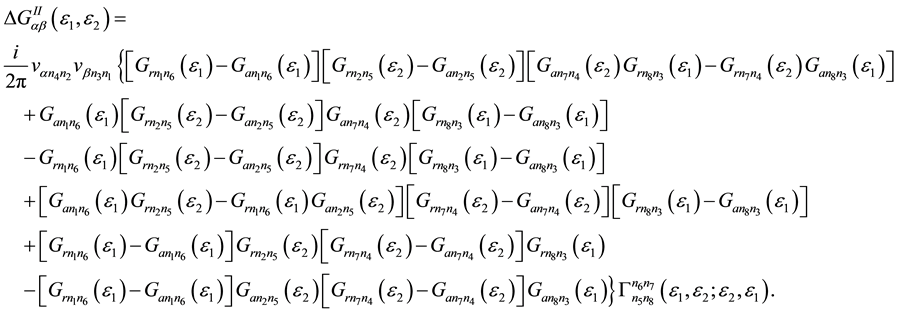 (2)
(2)
where 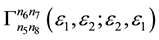 is vertex part of diagram for the mass operator of electron-electron interaction,
is vertex part of diagram for the mass operator of electron-electron interaction,
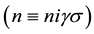 , here
, here 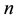 is number of primitive cell,
is number of primitive cell,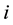 ―the sublattice-site number in primitive cell,
―the sublattice-site number in primitive cell,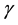 ―energy band index,
―energy band index,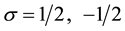 ―quantum number, which defines value of spin projection on z axis.
―quantum number, which defines value of spin projection on z axis.
Operator ―the projection of the electron velocity in (1) is:
―the projection of the electron velocity in (1) is:
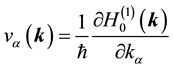 .
.
To simplify the formula (1) we use approximate expression for averaged value , where
, where ―is derived from the expression (2) by replacing the Green function
―is derived from the expression (2) by replacing the Green function  with Green function of effective medium
with Green function of effective medium .
.
In formula (1):
 (3)
(3)
where
 is scattering operator on the one site,
is scattering operator on the one site,
 is scattering operator on the two sites,
is scattering operator on the two sites,
 is coherent potential that describes effective medium,
is coherent potential that describes effective medium,
 ,
,  is atom sort and magnetic moment projection on z axis respectively.
is atom sort and magnetic moment projection on z axis respectively.
The value  in the expression for the single-center scattering operator
in the expression for the single-center scattering operator  that describes the scattering on potentials of ionic skeleton, static charge fluctuations and spin density is given by formula:
that describes the scattering on potentials of ionic skeleton, static charge fluctuations and spin density is given by formula:
 (4)
(4)
where ,
,  is number of electrons in
is number of electrons in  state for
state for  atom in crystal and effective medium
atom in crystal and effective medium
respectively,  is electron-electron interaction operator’s matrix element [3] -[5] .
is electron-electron interaction operator’s matrix element [3] -[5] .
In expression (1) values ,
, ―the probability of finding atoms of different sorts and values of localized magnetic moment projection at the lattice sites [3] -[5] . In the Hamiltonian of the system should be included term that is the operator of interaction energy of intrinsic magnetic moment of a particle with an external magnetic field.
―the probability of finding atoms of different sorts and values of localized magnetic moment projection at the lattice sites [3] -[5] . In the Hamiltonian of the system should be included term that is the operator of interaction energy of intrinsic magnetic moment of a particle with an external magnetic field.
Spin-dependent transport in systems with strong electronic correlations is described by partial constituent of diagonal conductivity tensor element (1), which corresponds to a specific value of the electron spin projection.
3. Results of Calculations and Conclusions
Here are the results of calculation of the energy spectrum of electrons and phonons and conductivity of carbon nanotubes doped with chromium. In calculation, renormalization of vertex parts of mass operator of electron-electron and electron-phonon interactions [3] -[5] was neglected. Real wave functions of 2s, 2p, 3s, 3d-states of neutral non-interacting atoms of carbon were chosen. The off-diagonal matrix elements  by site index of Hamiltonian calculated by taking into account the first three coordination spheres. Contribution to the static displacements of atoms and members was neglected in the calculations. Calculations were performed for the temperature T = 300 K.
by site index of Hamiltonian calculated by taking into account the first three coordination spheres. Contribution to the static displacements of atoms and members was neglected in the calculations. Calculations were performed for the temperature T = 300 K.
We performed geometry optimization of the crystal structure of carbon nanotube of chirality (3,0) with Cr impurity. Geometric optimization of the crystal structure was achieved by minimizing the free energy. Carbon nanotube doped with Cr has a one-dimensional crystal structure. Primitive cell contains 18 non-equivalent atom positions. Carbon atoms are located in 12 positions on the surface of the inner cylinder. The distance between the carbon atoms is 0.142 nm. Cr atoms are randomly located in the 6 position on the outer surface of the cylinder opposite the center of a hexagon, the vertices of which are carbon atoms. Through the study of free energy minimum found that Cr atoms are randomly located on the surface of nanotubes. This indicates that the probability of Cr atoms arrangement , where
, where ―the ratio of Cr atoms with respect to the six possible positions of the atoms Cr within the primitive cell. The distance between carbon atoms and Cr is 0.22 nm. The relative position of carbon atoms and Cr is similar to the location of atoms of transition metals on the surface of carbon nanotubes of large diameter, which are described in [1] [2] by ultrasoft pseudopotential method [6] [7] .
―the ratio of Cr atoms with respect to the six possible positions of the atoms Cr within the primitive cell. The distance between carbon atoms and Cr is 0.22 nm. The relative position of carbon atoms and Cr is similar to the location of atoms of transition metals on the surface of carbon nanotubes of large diameter, which are described in [1] [2] by ultrasoft pseudopotential method [6] [7] .
The value localized magnetic moment projection of the atom Cr and induced localized magnetic moment of an atom C in the direction of the magnetic field increases with the size of the field. For carbon nanotubes of 5 Cr atoms in primitive cell value projection magnetic moment of the atom Cr varies within , and the magnetic moment of the atom C―within
, and the magnetic moment of the atom C―within  with increasing values of the magnetic field from zero to
with increasing values of the magnetic field from zero to . The magnetic field is oriented along the axis of the carbon nanotube. Parameter of pair correlations in the orientation of localized magnetic moments on lattice sites for the first coordination sphere in the absence of magnetic field equals to
. The magnetic field is oriented along the axis of the carbon nanotube. Parameter of pair correlations in the orientation of localized magnetic moments on lattice sites for the first coordination sphere in the absence of magnetic field equals to . The value
. The value  for the second and third coordination spheres is close to zero. A positive value of
for the second and third coordination spheres is close to zero. A positive value of  for the first coordination sphere indicates that the localized magnetic moment given carbon atom is oriented in the same direction as the magnetic moment of the nearest Cr atom.
for the first coordination sphere indicates that the localized magnetic moment given carbon atom is oriented in the same direction as the magnetic moment of the nearest Cr atom.
Figure 1 shows a partial  and full
and full  densities of electron states
densities of electron states
of carbon nanotube with an admixture of Cr in the absence of external magnetic field. In the absence of a magnetic field . Vertical line shows the Fermi level
. Vertical line shows the Fermi level .
.
In Figure 2, points show the dependence of the free energy  on the parameter of pair correlations in the arrangement of Cr impurities on lattice sites
on the parameter of pair correlations in the arrangement of Cr impurities on lattice sites  for the first coordination sphere. Atom of Cr is denoted as atom of sort
for the first coordination sphere. Atom of Cr is denoted as atom of sort . The dependence
. The dependence  is shown in the region of free energy minimum. The free energy is measured from electrostatic interaction energy of ions of sort A.
is shown in the region of free energy minimum. The free energy is measured from electrostatic interaction energy of ions of sort A.
Figure 3 shows partial  and full
and full  densities of electron states of carbon nanotube with 5 atoms of Cr per primitive cell in external magnetic field
densities of electron states of carbon nanotube with 5 atoms of Cr per primitive cell in external magnetic field .
.
Figure 3 shows part of the energy spectrum that is close to the Fermi level.
As shown in Figure 3, for a given sign of the projection of localized magnetic moment on sites of lattice energies of the electron with spin  shifted relative to values of the energy of an electron with spin
shifted relative to values of the energy of an electron with spin . This leads to the formation of the Coulomb gap in the energy spectrum of electrons, which is visible on
. This leads to the formation of the Coulomb gap in the energy spectrum of electrons, which is visible on  of Figure 3. The results presented in Figure 3, qualitatively consistent with results obtained by another method in [2] .
of Figure 3. The results presented in Figure 3, qualitatively consistent with results obtained by another method in [2] .
In Figure 4, the dependence of the spin polarization electric current  of carbon nanotube with chirality (3,0) and 5 atoms of Cr per primitive cell on the magnitude of the external magnetic field calculated by the formula (1) for temperature 300 K is shown.
of carbon nanotube with chirality (3,0) and 5 atoms of Cr per primitive cell on the magnitude of the external magnetic field calculated by the formula (1) for temperature 300 K is shown.

Figure 1. Densities of electron states of carbon nanotube with an admixture of Cr.

Figure 2. Dependence of free energy F for carbon nanotubes with 5 atoms of Cr per primitive cell on parameter of pair correlations in the arrangement of Cr impurities on lattice sites .
.
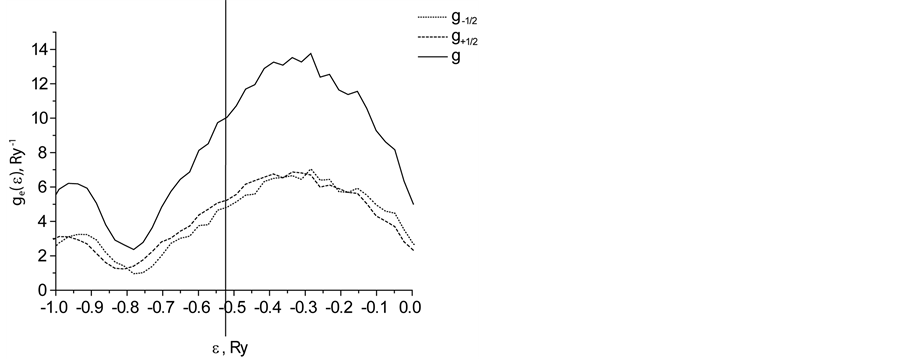
Figure 3. Densities of electron states of carbon nanotube with 5 atoms of Cr per primitive cell in external magnetic field .
.

Figure 4. The dependence of spin polarized electric current  of carbon nanotube on the magnitude of the external magnetic field
of carbon nanotube on the magnitude of the external magnetic field .
.
Thus, the phenomenon of spin-dependent electron transport in a carbon nanotube is the result of strong electron correlations caused by the presence of chromium atoms. The value of the spin polarization of electron transport determined by the difference of the partial densities of states (Figure 3) electrons with opposite spin projection at the Fermi level and the difference between the relaxation times arising from different occupation numbers of single-electron states  of atoms of carbon and chromium. The value of the spin polarization of the electric current increases with increasing of Cr atoms concentration and magnitude of the external magnetic field. In [1] calculated the electronic structure and properties of carbon nanotubes with transition metal chains, adsorbed on the surface, is based on density functional method using ultra-soft pseudopotential. Our results are qualitatively consistent with the results of [1] , in which ab-initio electron density functional method shows that the chains of transition metals adsorbed on the surface of carbon nanotubes, open a gap in the electrons states with a certain spin value.
of atoms of carbon and chromium. The value of the spin polarization of the electric current increases with increasing of Cr atoms concentration and magnitude of the external magnetic field. In [1] calculated the electronic structure and properties of carbon nanotubes with transition metal chains, adsorbed on the surface, is based on density functional method using ultra-soft pseudopotential. Our results are qualitatively consistent with the results of [1] , in which ab-initio electron density functional method shows that the chains of transition metals adsorbed on the surface of carbon nanotubes, open a gap in the electrons states with a certain spin value.
References
- Durgun, E. and Ciraci, S. (2006) Physical Review B, 74, Article ID: 125404. http://dx.doi.org/10.1103/PhysRevB.74.125404
- Yang, C., Zhao, J. and Lu, J.P. (2004) Nano Letters, 4, 561-563. http://dx.doi.org/10.1021/nl035104x
- Repetsky, S.P. and Shatnii, T.D. (2002) Theoretical and Mathematical Physics, 131, 832.
- Repetskii, S.P. and Vyshivanaya, I.G. (2005) Physics of Metals and Metallography, 99, 558.
- Repetskii, S.P., Vyshivanaya, I.G. and Cheshkovskii, D.K. (2012) Physics of Metals and Metallography, 113, 213-221. http://dx.doi.org/10.1134/S0031918X12030143
- Vanderbilt, D. (1990) Physical Review B, 41, 7892. http://dx.doi.org/10.1103/PhysRevB.41.7892
- Laasonen, K., Car, R., Lee, C. and Vanderbilt, D. (1991) Physical Review B, 43, 6796. http://dx.doi.org/10.1103/PhysRevB.43.6796