Advances in Bioscience and Biotechnology
Vol.05 No.10(2014), Article ID:50102,7 pages
10.4236/abb.2014.510098
A new tandem gene construction method involving a cloning system using Poxvirus DNA polymerase, and its application to gene expression
Tatsuro Shibui, Daisuke Sakaguchi, Hiroyoshi Hara
Food Biotechnology Laboratory, School of Food Sciences, Nippon Veterinary and Life Science University, Tokyo, Japan
Email: tshibui@nvlu.ac.jp
Copyright © 2014 by authors and Scientific Research Publishing Inc.
This work is licensed under the Creative Commons Attribution International License (CC BY).
http://creativecommons.org/licenses/by/4.0/



Received 15 July 2014; revised 24 August 2014; accepted 20 September 2014
ABSTRACT
A simple method for constructing polymerized genes using only restriction enzymes and commercially available cloning systems was established. In this system, gel isolations or purifications of target genes after restriction enzyme digestions or PCR amplifications, which often cause errors and mutations in the target gene sequence, are not necessary. To verify the usefulness of this me- thod, one, two, four, eight, and sixteen tandem-repeats of the Green Fluorescent Protein (GFP) ex- pression gene in Escherichia coli were sequentially constructed. Efficacies of the GFP gene expres- sion of those plasmids in E. coli showed an increasing trend in accordance with the copy numbers of the gene. On SDS polyacrylamide gel electrophoresis with Coomassie blue staining, no express- ed protein could be seen in E. coli cells harboring plasmids that contained one or two copies of the gene. However, expressed protein bands in E. coli cells were clearly detected with 4 copies of the gene. In quantitative analyses involving green fluorescence intensities per culture volume, the ex- pression level in E. coli with 16 copies of the gene was 36.3-fold higher than that in E. coli with one copy at 22 hours after induction.
Keywords:
Poxvirus DNA polymerase, gene cloning, tandem repeat, GFP, T7 RNA polymerase

1. Introduction
There have been many methods reported for the production of target proteins by genetic engineering. Typical systems usually involve bacteria [1] -[3] , yeast [3] - [6] , fungi [3] [7] , insect cells [8] , mammalian cells [9] , trans- genic insects [10] , animals [11] , and plants [12] - [14] . Those expression systems are basically built up with simi- lar genetic constructs, as follows:
1) Placing a transcriptional promoter with well-controlled and high-transcriptional activity at 5’ upstream, and a transcriptional terminator sequence to prevent transcriptional read-through at 3’ downstream of the target pro- tein’s genes [1] .
2) Inserting sequences to improve the translation of target genes, such as a 5’ cap structure, IRES (internal ri- bosome entry sites) [15] , SD (Shine-Dalgarno) sequence, etc., upstream of the gene [16] .
3) Optimizing codons of the target gene to improve translation efficiency in host cells [17] .
Besides those components, the gene-dosage is also important. The copy number of target genes often influ- ences the amount of target proteins produced in host cells [9] [18] .
Two methods have generally been reported for increasing the copy number of target genes, i.e., in vivo and in vitro methods. In in vivo methods, a DNA construct that contains target and drug-resistant marker genes is made, and inserted into host cells. Then, more drug-resistant clones that are deemed to harbor increased copy numbers of drug-resistant genes are selected. A few of those more drug-resistant clones also contain increased copy num- bers of target genes along with the multiplication of drug-resistant genes [9] . However, it remains uncertain whether or not we can always obtain clones that contain amplified target genes, and finding amplified clones of- ten requires simple assay methods for identification.
On the other hand, in vitro methods for increasing copy numbers of target genes in expression vectors are more reliable than in vivo ones, and have been typically described as follows: cutting a target gene and vector with restriction enzymes that generate the same cohesive or blunt ends, and separating the target gene from the vector, and then putting the target gene into a restriction site again with the same cohesive or blunt ends in the expression vector by ligation [18] . These methods are reliable ways to multiply the target gene in plasmid vectors by genetic engineering. However, they are time-consuming and laborious.
In this report, we describe a simple new method for increasing copy numbers of a target gene in an expression vector in vitro using commercially available cloning kits, which could be applied for many recombinant protein production systems as well as genetic reorganizations.
2. Materials and Methods
2.1. Bacterial strains
JM 109 competent cells were purchased from Toyobo Biochemicals (Japan) and used for the construction of plasmids. BL21 (DE3) competent cells were purchased from BioDynamics Laboratory Inc. (Japan), and used for protein expression.
2.2. Media
LB agar medium (Life Technology, USA) supplemented with Kanamycin (Meiji Pharmaceuticals) at 15 mg/L (LB agar Km) was used for the construction of plasmids and expression experiments. M9 Minimal Salt medium containing 2% Casamino acid, 1 mM CaCl2, 1 mM MgSO4, 1% glucose, and Kanamycin at 15 mg/L (M9 Km) was used for expression experiments in liquidcultures.
2.3. Reagents
Restriction enzymes were purchased from New England Biolabs (USA). Iso-propyl-β-D-galactopyranoside (IPTG) was from Sigma (USA).
An expression vector, pET28a, was from Novagen (USA). A plasmid, pEU-E01, containing the Green Fluo- rescent Protein (GFP) gene was described previously [19] .
Oligo DNAs used in this study are listed. They were ordered from and synthesized by Medical & Biological Laboratories Co., LTD. (Japan).
The cloning kit, which uses unique properties of the 3’→5’ exonuclease activity of Poxvirus DNA polyme- rase [20] , was from Clonetech (Infusion cloning kit, USA), and used according to the manufacturer’s protocol. PCR was performed with hi-fidelity DNA polymerase (Prime Max PCR Kit, Clonetech, USA), according to the supplier’s manual. DNA purification kits were purchased from Nippon Genetics, Gbm (Japan) (Table 1).
 (a)(b)
(a)(b)
Table 1. A list of synthetic DNAs.
1)&3)The underlined sequences are the Shine-Dalgarno sequence, and the ATG sequences (bold letters) are the start codon of the GFP gene. The italic sequence is TSL. 2)The italic sequence is TSL/R BH, and the TTA sequence (bold letters) is the termination codon of the GFP gene.
pET 28a was completely digested with Nco I and Xho I, and purified with the DNA purification kit. The puri- fied GFP expression unit for E. coli was mixed with the linearized vector, and joined by the cloning system us- ing Poxvirus DNA polymerase. JM 109 competent cells were transformed with the reaction mixture, and se- lected on LB agar Km. Plasmids in two transformants had their insert checked by digestion with Nco I and Xho I. All clones showed correct insertion of the GFP gene. The plasmid pETTSLR (GFP)1 was prepared from one of the clones with a plasmid midi prep. kit (Nippon Genetics, Japan).
2.5. Construction of plasmids That contain tandem-repeated GFP expression units
pETTSLR (GFP)1 was completely digested with Eco R V and Bam H I, independently. Each digest was purified by the DNA purification kit, mixed, and joined with the cloning kit using Poxvirus DNA polymerase to construct a plasmid, pETTSLR (GFP)2, with the insertion of two repeated GFP expression units. JM 109 was transformed by the reaction mixture, and selected on LB Km. The transformants had their plasmids checked by Eco R V di- gestion to obtain pETTSLR (GFP)2.
In the same manner, pETTSLR (GFP)2 was digested with Eco R V and Bam H I separately, and they were then mixed and treated with the cloning kit to construct a plasmid, pETTSLR (GFP)4, containing four tandem- repeated GFP expression units. Thus, we sequentially constructed the plasmids pETTSLR (GFP)8 from pETTSLR (GFP)4 and pETTSLR (GFP)16 from pETTSLR (GFP)8, containing eight and sixteen tandem-repeated GFP expression units, respectively.
2.6. Expression of GFP
The plasmids constructed in the previous section were introduced into BL21 (DE3) to produce GFP. BL21 (DE3) harboring those plasmids was cultured overnight in 1 mL of LB medium supplemented with 15 mg/mL of Ka- namicin at 37˚C. Aliquots of 0.05 mL of the cultures were streaked on a paper filter (a coffee filter) on an LB agar Km plate, and incubated at 30˚C for 2 hours. Then, the filter was transferred to an LB agar Km plate sup- plemented with 1 mM IPTG for the induction of the GFP gene, and incubated at 30˚C for 22 hours. The expres- sion was monitored bygreen fluorescence from the GFP produced in E. coli harboring each plasmid. Green flu- orescence of E. coli cells emitted on irradiation with a blue light LED (L = 550 nm) (Biocraft, Japan) was de- tected through an orange filter, and photos were taken at 3 and 22 hours after induction.
2.7. GFP production levels monitored by sodium dodecyl sulfate (SDS) polyacyl amide gel electrophoresis
E. coli cells possessing each plasmid were cultured overnight in M9 km at 30˚C. Aliquots of 0.1 mL were used to inoculate 10 mL of fresh M9 Km in flasks, and incubated at 30˚C with rotary shaking at 230 rounds per minute (r.p.m). After 2 hours, IPTG was added at a final concentration of 1 mM for induction. At 3 and 22 hours after induction, cells were harvested by centrifugation at 5000 r.p.m. for 10 minutes at 4˚C. The supernatants were removed, and cell pellets were stored at −20˚C until use. Those time-points were selected based on data from the previous Section 2.6.
Cells from 10 mL culture were resolved in a sample buffer, and incubated at 98˚C for 10 minutes. Then, the samples underwent SDS-15% polyacrylamide gel electrophoresis (PAGE). The proteins were stained by Coo- massie Brilliant Blue (Wako Chemicals Corporation, Japan).
2.8. Measuring green fluorescent intensity in cell extracts
E. coli cell pellets prepared and stored in the previous section were suspended in 1/10 culture volumes of PBS (10 mM Sodium phosphate, pH 7.5, and 150 mM NaCl), and cells were lysed by ten freeze-and-thaw cycles. Cell supernatants were prepared by removing the cell debris with centrifugation at 15,000 r.p.m. for 10 minutes. The relative fluorescent activity of GFP in each cell lysate was measured with emission light at 535 nm caused by excitation light at 485 nm using a fluorescence spectrophotometer (ARVO MX, Perkin Elmer). All fluorescence spectra were analyzed after a 10 to 100-fold dilution of the supernatants with PBS, and measured in triplicate.
3. Results and Discussion
3.1. The sequences required for creating tandem-repeated genes
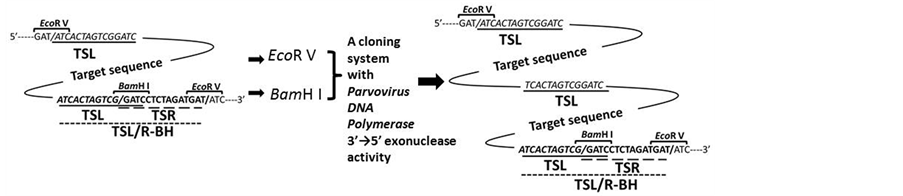
The cloning kit used in this study is ligation-independent, using the unique properties of the 3’ → 5’ exonuc- lease activity of Poxvirus DNA polymerase [20] . When incubated with linear duplex DNAs with homologous ends, the 3’ → 5’ proofreading activity of Poxvirus DNA polymerase progressively removes nucleotides from the 3’ end in the presence of Mg2+ and low concentrations of dNTPs. Complementary regions of substrate DNAs that can then spontaneously anneal through base pairing are exposed. This results in joined molecules containing a hybrid region flanked by nicks, gaps, or short overhangs. Their introduction into E. coli repairs any single- stranded gaps. Thus, one copy of the overlap is present in the final DNA product.
Considering the above characteristics of the cloning systems, we designed a sequence that could make target sequences tandem without PCR amplification or separation of the fragment from vectors. An example sequence for this purpose is illustrated in Figure 1(a). A sequence recognized by Eco R V was chosen for separation of the target genes from their vector. In order to mimic the conditions described in the cloning kit’s protocol, we selected Eco R V, since digestion with Eco R V produces blunt-ended DNA fragments similar to the ones am- plified by PCR. Any sequences recognized by restriction enzymes that could generate blunt-ended DNA frag- ments upon digestion could be used for this purpose, unless they do not exist in the target gene. Since the clon- ing system requires 15 base-pair (bp) homologous sequences at both ends of the target DNA fragment and the cloning vector, we put TSL and TSLR sequences at both ends of the target DNA. In this study, we chose Bam H I for cloning the fragment in the vector. Thus, the 3’ end sequence of the target gene was designed as TSLR/BH, shown in Figure 1. Since the cloning systems with Poxvirus DNA polymerase 3’ → 5’ exonuclease activity use 15 bp homologous sequences, the cloning systems excel in the directional cloning of the target genes compared to traditional cloning systems using ligase.
3.2. Polymerization of target gene
With Eco R V and Bam H I digestions, and a cloning system using Poxvirus DNA polymerase 3’ → 5’ exonuc- learase activity, pluralized genes could be sequentially constructed in a vector containing TSLR/BH, as shown in Figure 1. In our designed sequence, since there is always one Bam H I site in vectors, any DNA fragments that have TSL at 5’ and TSLR BH at 3’ ends could be inserted into the Bam H I site in the vector. The limit of the copy number of the pluralized target gene is considered to depend on the DNA-uptake ability of competent E. coli cells. We found it difficult to obtain transformed cells when plasmids were over 25 kbp (data not shown). In our GFP expression unit, we could produce sixteen repeats of the expression unit. Since the length of 16 repeated GFP expression units is 12.4 kbp and that of pET28a is 5.1 kbp (see Figure 2), we concluded that 16 copies in the expression vector (total: 17.5 kbp) is almost the maximum size for the transformation of chemical- or electro-competent E. coli cells. On using bacteriophage packaging systems like λ and cos (λ phage packaging sequence) to introduce the desired plasmids, plasmids that contain higher copy-number insertions than using typical competent cells can be
 (a)
(a)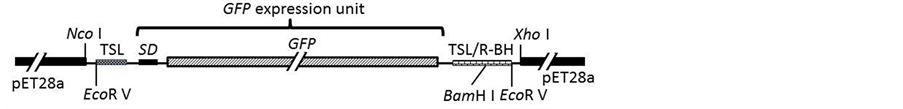 (b)
(b)
Figure 1. A nucleotide sequence designed for the construction of repeated genes, and its application for a GFP expression unit in E. coli. (a) Illustration of a target gene sandwiched in the designed sequence. TSL and TSR indicate 5’ and 3’ end-overlapped sequences (15 bp) for creating tandem genes, respectively; (b) A GFP expression unit flanked by TSL and TSL/R- BH in an expression vector, pET28a. TSL and TSR: Left and right end-sequences required for construction of the tandem-repeated gene, respectively. TSL/R-BH: A cloning site-sequence of the target gene with Bam H I digestion. SD: Shine-Dalgarno (ribosome binding) sequence. GFP: Green fluorescent protein gene.

Figure 2. Agarose gel electrophoresis analysis of the constructed plasmids. pETTSLR (GFP)1, 2, 4, 8, and 16 were digested with Eco R V, and analyzed by 1% agarose gel electrophoresis. Arrows A, B, C, D, and E indicate the GFP expression unit DNA fragments in each plasmid and 1, 2, 4, 8, and 16 copies of the unit DNA, respectively.
obtained. Alkali-SDS methods for the isolation of plasmids also tend to work inefficiently with plasmids over 15 kbp.
rec A mutant E. coli hosts, like JM109, are also important for the stable construction of plasmids with tan- dem-repeat sequences. We could not obtain plasmids with tandem-repeat sequences in rec A wild-type strains, like MC1061. However, the reason is unclear. Once these plasmids had been constructed in rec A mutant strains, they could be introduced and stably maintained in rec A wild-type strains. Thus, we were able to use the rec A wild-type strain, BL21 (DE3), in expression experiments.
Before joining the fragment and vector using the kit, the digested fragment and vector were separately puri- fied with a DNA purification kit using a silica gel membrane. When we mixed the two digests and purified them together in one column in order to reduce time and cost, and then joined them using the kit, no colonies appear- ed in transformation. The homologous sequences may interact with each other upon purification before being treated with the cloning kit, and this may have adverse effects on the kit’s reaction.
3.3. Effects of polymerized expression units on protein production
Expression units in the expression vectors are not transcribed in JM109, since a T7 promoter in the expression vector controls their transcriptions, and JM109 does not contain a T7 RNA polymerase gene. Hence, those plas- mids containing the GFP expression units were introduced into BL21(DE3), which possesses a T7 RNA polyme- rase gene whose expression is under the control of the lacL8UV5 promoter, and induced by adding isopropyl- β-D-galactopyranoside (IPTG) to the medium.
The expression levels of GFP were firstly monitored by culture on an LB Km plate using a blue light LED. The levels increased according to the copy number of the expression unit with IPTG induction (Figure 3). The expression showed toxicity for E. coli cells: when we streaked cells directly on the LB Km plate containing IPTG instead of prior incubation for 3 hours without IPTG, no cells grew on the plate.
The protein expression levels in M9 Km liquid cultures were also checked by SDSPAGE and a fluorescence spectrophotometer. Although the expressed protein band could not be observed in cells with 1 or 2 copies of the units by Coomassie staining of the gel, it was recognized with4 copies of the expression unit (Figure 4). With this tandem gene method, even if expressed protein bands cannot be observed using a plasmid with one copy insert by Coomassie staining, strains producing sufficient levels of the target protein for detection by Coomassie staining can be obtained.
The expression level was also analyzed by fluorescence spectrophotometry. E. coli harboring 4, 8, and 16 copies of the GFP expression unit showed high fluorescence intensities per culture volume (Figure 5). In E. coli cells with 16 copies, the fluorescence intensity reached a level 36.3-times higher than that of one-copy holders at 22 hours after induction.
4. Conclusions
· A sequence for the construction of tandem-repeat genes by a cloning system using Poxvirus DNA polyme- rase 3’ → 5’ nuclease activity was designed.

Figure 3. GFP expression in BL21 (DE3) harboring pETTSLR (GFP)1, 2, 4, 8, and 16. C: Negative control (pET28a), 1: pETTSLR (GFP)1, 2: pETTSLR (GFP)2, 3: pETTSLR (GFP)4, 4: pETTSLR (GFP)8, 5: pETTSLR (GFP)16. Cells incubated at 30˚C for 3 hours after in- duction. Cells incubated at 30˚C for 22 hours after induction. Cells were irradiated with a blue LED (550-nm wavelength), and their fluorescence was recorded with a digital camera through an orange filter.

Figure 4. SDS-PAGE patterns of BL21(DE3) harboring pETTSLR (GFP)1, 2, 4, 8, and 16. M: Molecular weight markers, C: Negative control (pETTSLR (GFP)1 without induction), 1: pETTSLR (GFP)1, 2: pETTSLR (GFP)2, 3: pETTSLR (GFP)4, 4: pETTSLR (GFP)8, 5: pETTSLR (GFP)16. In 1 - 5, cells were cultured in M9 Km at 30˚C for 22 hours after in- duction by 1 mM IPTG. Cells from 10-μLculture underwent 15% SDS-PAGE. The gel was stained with Coomassie blue. The arrow indicates the GFP position.

Figure 5. Relative fluorescent intensities of BL21(DE3) harboring pETTSLR (GFP)1, 2, 4, 8, and 16. □: Cells were cultured in M9 Km at 30˚C without induction. ■: Cells were cultured in M9 Km at 30˚C for 3 or 22 hours after induction by 1 mM IPTG. Cell lysate was prepared and fluorescent intensities were measured as described in Materials and Methods.
· The sequence worked well for the construction of tandem-repeat genes.
· With an expression gene for GFP, up to 16 copies of the gene were successfully introduced into the pET28a vector.
· The expression levels of GFP in E. coli harboring vectors increased in accordance with the copy number of the expression gene in the vector.
· E. coli cells containing plasmids with 16 tandem-repeats of the expression gene produced 36.3-fold higher levels of GFP than those with one copy of the gene in M9 liquid culture for 22 hours under an induced con- dition.
References
- Shibui, T. and Nagahari, K. (1992) Secretion of a Functional Fab Fragment in Escherichia coli and the Influence of Culture Conditions. Applied Microbiology and Biotechnology, 37, 352-357. http://dx.doi.org/10.1007/BF00210991
- Dolgikh, V.V., et al. (2014) Optimization of the Protocol for the Isolation and Refolding of the Extracellular Domain of HER2 Expressed in Escherichia coli. Acta Naturae, 6, 106-109.
- Rosano, G.L. and Ceccarelli, E.A. (2014) Recombinant Protein Expression in Microbial Systems. Frontiers in Microbiology, 5, 1-2.
- Goodrick, J.C., et al. (2001) High-Level Expression and Stabilization of Recombinant Human Chitinase Produced in a Continuous Constitutive Pichia pastoris Expression System. Biotechnology and Bioengineering, 74, 492-497. http://dx.doi.org/10.1002/bit.1140
- Farhadi, B., Shekari Khaniani, M. and Mansoori Derakhshan, S. (2014) Construction of pPIC9 Recombinant Vector Containing Human Stem Cell Factor. Advanced Pharmaceutical Bulletin, 3, 303-308.
- Fazel, R., et al. (2014) Cloning and Expression of Aspergillus flavus Urate Oxidase in Pichia pastoris. SpringerPlus, 3, 395.
- Nevalainen, K.M., Te’o, V.S. and Bergquist, P.L. (2005) Heterologous Protein Expression in Filamentous Fungi. Trends in Biotechnology, 23, 468-474. http://dx.doi.org/10.1016/j.tibtech.2005.06.002
- Caron, A.W., Archambault, J. and Massie, B. (1990) High-Level Recombinant Protein Production in Bioreactors Using the Baculovirus-Insect Cell Expression System. Biotechnology and Bioengineering, 36, 1133-1140. http://dx.doi.org/10.1002/bit.260361108
- Kito, M., et al. (2002) Construction of Engineered CHO Strains for High-Level Production of Recombinant Proteins. Applied Microbiology and Biotechnology, 60, 442-448. http://dx.doi.org/10.1007/s00253-002-1134-1
- Aflakiyan, S., et al. (2013) Expression of the Recombinant Plasminogen Activator (Reteplase) by a Non-Lytic Insect Cell Expression System. Research in Pharmaceutical Sciences, 8, 9-15.
- Yu, H., et al. (2013) Large-Scale Production of Functional Human Lysozyme in Transgenic Cloned Goats. Journal of Biotechnology, 168, 676-683.
- Fussenegger, M. and Hauser, H. (2007) Protein Expression by Engineering of Yeast, Plant and Animal Cells. Current Opinion in Biotechnology, 18, 385-386. http://dx.doi.org/10.1016/j.copbio.2007.10.002
- Strasser, R., Altmann, F. and Steinkellner, H. (2014) Controlled Glycosylation of Plant-Produced Recombinant Pro- teins. Current Opinion in Biotechnology, 30C, 95-100.
- Tuse, D., Tu, T. and McDonald, K.A. (2014) Manufacturing Economics of Plant-Made Biologics: Case Studies in Therapeutic and Industrial Enzymes. BioMed Research International, 2014, Article ID: 256135.
- Somers, J., Poyry, T. and Willis, A.E. (2013) A Perspective on Mammalian Upstream Open Reading Frame Function. The International Journal of Biochemistry & Cell Biology, 45, 1690-1700. http://dx.doi.org/10.1016/j.biocel.2013.04.020
- Shibui, T. and Nagahari, K. (1994) Antibody Produced by Using Escherichia coli Expression Systems. Food and Bio- process Technology, 19, 253-268.
- Patterson, S.S., et al. (2005) Codon Optimization of Bacterial Luciferase (Lux) for Expression in Mammalian Cells. Journal of Industrial Microbiology Biotechnology, 32, 115-123. http://dx.doi.org/10.1007/s10295-005-0211-8
- Shibui, T., et al. (1991) High-Level Secretion of Human Apolipoprotein E Produced in Escherichia coli: Use of a Se- cretion Plasmid Containing Tandemly Polymerized ompF-Hybrid Gene. Journal of Biotechnology, 17, 109-120. http://dx.doi.org/10.1016/0168-1656(91)90002-D
- Koga, H., Misawa, S. and Shibui, T. (2009) A Wheat Embryo Cell-Free Protein Synthesis System Not Requiring an Exogenous Supply of GTP. Biotechnology Progress, 25, 1322-1327. http://dx.doi.org/10.1002/btpr.230
- Zhu, B., et al. (2007) In-Fusion Assembly: Seamless Engineering of Multidomain Fusion Proteins, Modular Vectors, and Mutations. BioTechniques, 43, 354-359. http://dx.doi.org/10.2144/000112536