Advances in Bioscience and Biotechnology
Vol.5 No.3(2014), Article ID:42622,4 pages DOI:10.4236/abb.2014.53025
Malignant hyperthermia: A runaway thermogenic futile cycle at the sodium channel level
![]()
100 Winterset Ct., Sunrise Beach, MO 65079, USA
Email: chwilliams2135@sbcglobal.net
Copyright © 2014 Charles H. Williams. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited. In accordance of the Creative Commons Attribution License all Copyrights © 2014 are reserved for SCIRP and the owner of the intellectual property Charles H. Williams. All Copyright © 2014 are guarded by law and by SCIRP as a guardian.
Received 11 December 2013; revised 11 January 2014; accepted 24 January 2014
Keywords: Malignant Hyperthermia; Thermogenic
ABSTRACT
Malignant Hyperthermia (“MH”)—the rapid onset of extremely high fever with muscle rigidity—is caused by a runaway heat production futile cycle mediated via the sodium channels at the myoneural receptor sites. MH is not triggered by non-depolarizing muscle relaxants; however, depolarizing muscle relaxants may trigger it [1]. Here we present a de novo hypothesis of how MH is triggered and develops. We believe that the acetylcholine receptor/sodium channels in the muscles of MH susceptible pigs initiate MH by allowing an increased flux of sodium ions when it is depolarized by acetylcholine or other depolarizing agents, such as succinylcholine and Halothane. Our theory is consistent with our observations of the effects of general anesthetics over twenty years. Succinylcholine is a depolarizing agent that is a potent MH trigger. Acetylcholine, the natural depolarizing muscle activator, may trigger MH if the susceptible patient or animal is exposed to sufficient stress, i.e., during strenuous activity, such as transport, fighting, breeding, etc. Halothane apparently destabilizes the myoneural sodium channels, which rapidly induces MH. The increased sodium channel activity releases heat with cascades that further releases of heat which results in the rapid onset of MH. MH susceptible pigs have increased action potential amplitudes at their myoneural junctions that are abnormally long in duration. This increased activity is thought to induce hypertrophy of muscle mass, increase metabolic rate, and cause other physical manifestations. When slaughtered, this increased metabolic activity causes the rapid post mortem release of heat in the muscles of MH susceptible pigs and, at the same time, the accumulation of low acidity, all of which denatures the muscle proteins to result in a pale, soft, exudative, pork meat considered to be of lesser quality for human consumption. The potency of inhalation anesthetics as a MH triggers varies widely. The inhalation anesthetic Halothane is a strong trigger of MH, causing MH within minutes of exposure. In contrast, the anesthetic Sevoflurane is a very weak trigger of MH, requiring several hours of inhalation exposure to trigger MH. Because of this, changing from Halothane to Sevoflurane as the general anesthetic of choice for surgeries in hospitals in the Greater Kansas City area during 1994 to 2006 led to an 11-fold decrease in the incidence of MH, from 1:50,000 to 1:550,000 [11]. One non-depolarizing muscle relaxant, Organon 9426 (“Rocuronium”) temporarily prevents MH in MH susceptible pigs when they are given sufficient dosages of it before being challenged with either Halothane or succinylcholine. Binding Rocuronium to the myoneural receptor sites apparently stabilizes them, thereby preventing increased sodium channel activity, and resulting MH. However, other non-depolarizing muscle relaxants do not have this protective effect— for examples Vecuronium, Arduan, and Organon 9616 do not. Uncoupling of mitochondria is not the source of accelerated heat production in MH susceptible pigs, as heart, liver, and skeletal muscle mitochondria isolated from MH susceptible pigs are all competent. Malignant Hyperthermia; Thermogenic
1. INTRODUCTION
Our population of MH susceptible pigs began in 1969. We conducted selective breeding and culling over 20 years for genetic studies and to produce highly MH susceptible pigs for use in experiments. We isolated heart, skeletal muscle, and liver mitochondria from MH susceptible pigs and found all were tightly coupled and exhibited normal metabolic rates without abnormalities.
We started our first series of experiments in 1970 with muscle relaxants. We observed that neither pancuronium nor curare, both non-depolarizing muscle relaxants, triggered MH in MH susceptible pigs; however, both succinylcholine (a depolarizing muscle relaxant) and Halothane were potent triggers.
Next, Hoech et al. performed a series of experiments challenging MH susceptible pigs with Metubine Iodide, a non-depolarizing muscle relaxant derivative of curare. It did not trigger MH. However, if the pigs were challenged with Halothane after administration of the Metubine Iodide, the Halothane triggered MH, i.e., the Metubine Iodide did not protect them from Halothane induced MH [2]. However the Metubine Iodide did protect the the pigs if a pharmacologic dose of the metubine iodide was administered [2].
Later, Bowman et al. measured the action potential at the myoneural junction in conscious, but untreated MH susceptible and control pigs, and found the action potential to be both significantly higher in voltage and longer in duration than normal. “The values were 300 µV and 3.2 ms respectively in the control pigs and 555 µV and 3.6 ms respectively in the young MHS [MH susceptible] pigs. These data were significantly larger at P < 0.01 and longer P < 0.05 respectively than the MUP [motor unit potential] of control pigs.” This was the first indication of an abnormality at the myoneural junction of MH susceptible pigs [3].
Subsequent experiments at TTUHSC, El Paso [4] in 1983 with the non-depolarizing muscle relaxant Vecuronium showed that it did not trigger MH in MH susceptible pigs and that it did not protect them from Halothane induced MH. Further experiments were conducted with the non-depolarizing muscle relaxants, Arduan, Organon 9616, and Organon 9426 (“Rocuronium”). These muscle relaxants did not trigger MH in MH susceptible pigs; however, unlike Arduan, Organon 9616, and Metubine Iodide, Organon 9426 prevented Halothane induced MH in MH susceptible pigs when they were exposed to Halothane while still under the influence of Organon 9426.
A linear regression analysis of the dose response data for Organon 9426 in MH susceptible pigs indicates that 427.033 µg/kg would be required to produce a 100% neuromuscular block. This is 73% of the amount of Organon 9426 required to produce a 100% neuromuscular block (586.31 µg/kg) in control pigs.
These experimental observations have led us to a de novo hypothesis of how MH is triggered and develops. We believe that the acetylcholine receptor/sodium channels in the muscles of MH susceptible pigs initiates MH by allowing an increased flux of sodium ions when it is depolarized by acetylcholine and/or succinylcholine. The succinylcholine initially depolarizes the myoneural junction then blocks it, which causes paralysis. In MH susceptible pigs the increased sodium flux is a runaway futile cycle that releases heat which synergistically accelerates the release and flow of sodium ions via the sodium channels. This effect is similar to a short circuit in a battery which increases electrical flow and produces heat.
Secondarily, muscle rigor develops, and blood flow, blood pressure, lactate production, carbon dioxide production, norepinephrine release all increase. Further, intense peripheral vasoconstriction develops, and metabolic rates rise greater than 10-fold, with concomitant core temperatures increases of up to 118˚ F (48˚C) [5,6].
Post mortem metabolic changes in slaughtered MH susceptible pigs cause high muscle temperatures, the rapid development of acidic pH levels and the denaturation of the muscle proteins which results in a pale, soft, exudative, pork meat of lesser quality for carcass salability.
Our testing of Sevoflurane anesthesia in 1984 led to the human clinical use of Sevoflurane, which resulted in a subsequent decrease in the incidence of MH in hospitals in the Greater Kansas City Area from 1:50,000 to 1:550,000. Sevoflurane is a very weak MH trigger, requiring two to three hours of exposure to trigger MH in MH susceptible pigs. In contrast, Halothane can trigger MH in them as quickly as one to three minutes. Halothane apparently destabilizes the myoneural sodium channels and causes muscle tremor as it reaches the muscle through the blood stream [6].
2. SUMMARY AND CONCLUSIONS
In MH susceptible pigs a genetic defect in the sodium channel protein in the muscle allows an excess of sodium ions to flow into the muscle cell. This genetic defect manifests itself in a higher than normal, and longer in duration, action potential at the Motor Unit Potential.
Acetylcholine release from transportation stress, breeding activity, fighting, hot weather, and other environmental stresses on animals can activate the MUP, and in MH susceptible pigs may cause the rapid onset of MH. (Figure 1).
Succinylcholine can cause MH in MH susceptible pigs by depolarizing the sodium channel and keeping it open for longer than normal periods of time. Halothane anesthesia causes a twitching response in the muscles of MH susceptible pigs which indicates activation of the MUP to open the sodium channels. This is probably how Halothane triggers MH.
Non-depolarizing muscle relaxants:
Pancuronium, Metubine iodide, Vecuronium, Pipecurium, Atracurium, and Organon 9426 (Rocuronium), are all non-depolarizing muscle relaxants, and do not trigger MH. If administered in sufficient dosage, Organon 9426
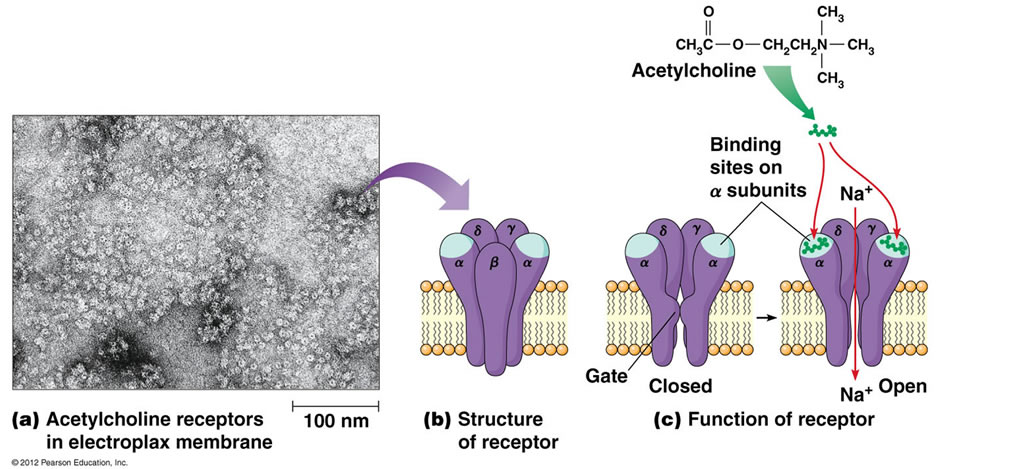
Figure 1. Acetylcholine-sodium channel function at the membrane level (from Wikipedia).
temporarily prevents MH in MH susceptible pigs, even after a 100% twitch response has returned after a blocking dose of Organon 9426. Recovering the animals, holding them for one week for drug washout, then administering Halothane again triggered MH in them, showing the prophylactic effect of Organon 9426 is temporary.
Selective breeding and culling for the MH gene produced a population of MH susceptible pigs that were highly susceptible to MH triggering agents. We suspect that selective line-breeding concentrates the defective sodium channel subunits into the sodium channels and thereby makes the sodium channels leaky. The leaky sodium channels then makes the animals susceptible to any factor that activates the depolarization response.
Further, leaky sodium channels provide a mechanism for muscle to continue heat production at a high rate post mortem and thereby activate glycolysis to produce lactate, with the resultant denaturation of muscle proteins observed as pale, soft, exudative pork meat with decreased carcass quality for marketability.
We suspect that the MH gene was selected for in pigs during the 1950’s when thin back fat was used as a sole selection criteria for breeding and marketing. During the 1950’s and 1960’s several animal science groups and meat laboratories were actively involved in researching stress and meat quality problems in pigs [7,8].
We used our population of MH susceptible pigs for over 20 years; they are an outstanding animal model for human MH [1-5,7-13].
Leaky sodium channels allow sodium ions (Na+) to flow into the muscle cell at higher than normal rates. This activates the Sodium-Potassium pump with results in a rapid consumption of ATP which, in turn, produces ADP plus Pi and heat (Figure 2). The released heat in-
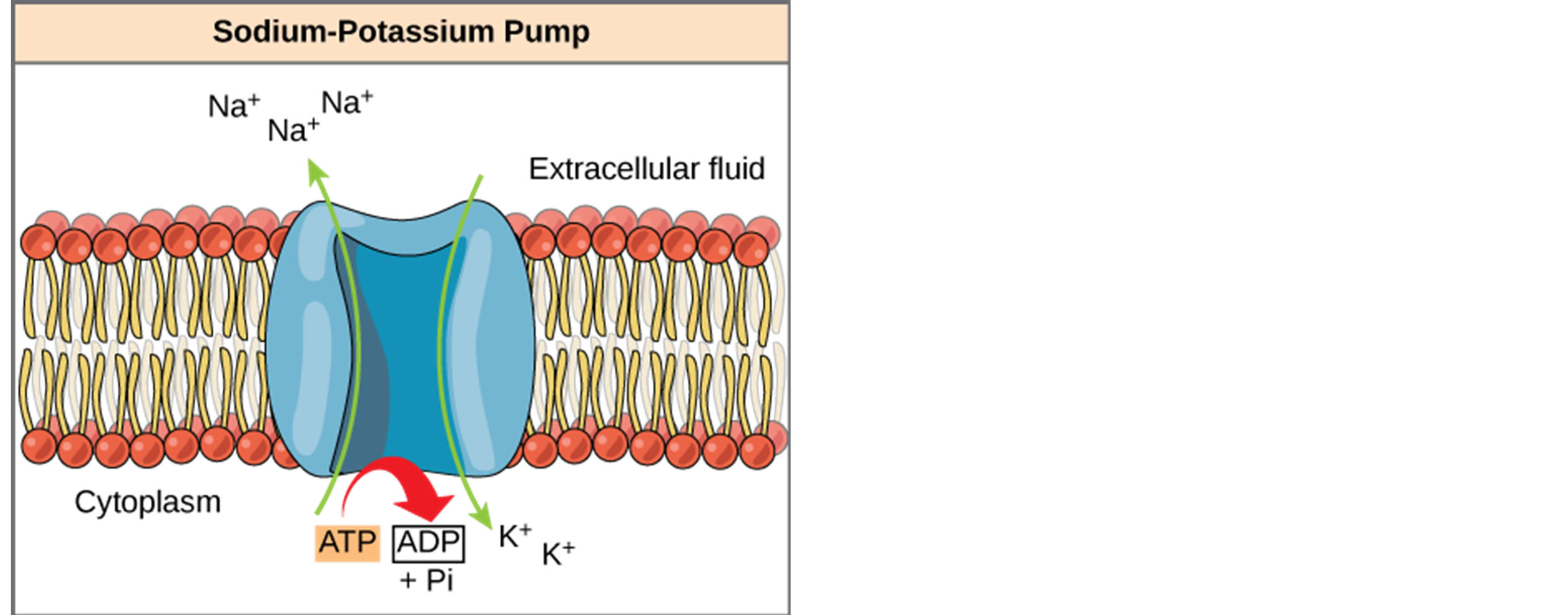
Figure 2. Sodium-Potassium pump which uses ATP to pump sodium ions out of the cell. The hydrolysis of ATP to ADP + Pi also releases heat at approximately 60% of the delta G −50 kJ/mol (from Wikipedia).
creases the activity of the thermogenic futile cycle increasing the overall metabolic rate as much as 10 to 15 times normal. This overall metabolic rate increase activates mitochondrial activity and glycolitic activity to resupply ATP at higher rates.
The testing of Sevoflurane in MH susceptible pigs has led to the widespread use of a new anesthetic that is safer to use in human patients, and is an exemplary example of the use of an animal model for a human condition.
Succlinylcholine is a potent triggering agent for MH and its use on humans should stop. The case of Stephanie Kuleba in West Palm Beach, Florida, is the latest example for why the use of succinylcholine should end.
Our de novo hypothesis for the biochemistry of MH development fits all the experimental facts, and will enable further rational testing of the proteins involved in the defective sodium channels. While this thermogenic futile cycle appears to be the primary mechanism for normal body heat production in normal individuals, it appears that due to a genetic defect in the sodium channel proteins it functions in an aberrant way in MH susceptible individuals which may lead to a lethal episode of MH.
ACKNOWLEDGEMENTS
I thank Nathan C Williams, Steven and Rachelle Gegg, for editing and revising the manuscript.
REFERENCES
- Williams, C.H. (2014) Malignant hyperthermia: A runaway futile cycle at the sodium channel level. Experimental Biology, San Diego, April in press.
- Hoech, G.P., Roberts, J.T., Williams, C.H., Waldman, S.D., Simpson, S.T., Trim, C.M. and Brazile, J. (1979) Prevention of porcine malignant hyperthermia with metocurine. In: Thermoregulatory mechanisms and their therapeutic implications. 4th International Symposium on the Pharmacology of Thermoregulation at Oxford, pp. 137- 141: Karger, Basel: 1980.
- Steiss, J.E., Bowman, J.M. and Williams, C.H. (1981) Electromyographic evaluation of malignant hyperthermia-susceptible pigs. American Journal of Veterinary Research, 42, 1173-1176.
- Buzello, W., Williams, C.H., Chandra, P., Watkins, M.L. and Dozier, S.E. (1985) Vecuronium and porcine malignant hyperthermia. Anesthesia and Analgesia, 64, 515- 519.
- Williams, C.H., Houchins, C. and Shanklin, M.D. (1975) Pigs susceptible to energy metabolism in the fulminant hyperthermia stress syndrome. British Medical Journal, 3, 411-413. http://dx.doi.org/10.1136/bmj.3.5980.411
- Williams, C.H., et al. (1988) Cardiac performance and hemodynamics in malignant hyperthermia susceptible and normal pigs during sevoflurane anesthesia. New York State Society of Anesthesiologists Meeting, NY Hilton.
- Harper, A. (2004) Top quality commercial market hogs and champion show pigs, are they the same. Part 2 Livestock update, JuneVA cooperative extension.
- Lynch, G. (1967) Ultrasonic and direct measurement of backfat thickness bacon pigs. Irish Journal of Agricultural & Food Research, 6, 41-47.
- Clark, M.G., Williams, C.H., Pfiefer, W.F., et al. (1973) Accelerated substrate cycling of fructose-6-phosphate in the muscle of malignant hyperthermic pigs. Nature, 245, 99-101.
- Williams, C.H. (1988) Experimental Malignant Hyperthermia. Springer Verlag, Berlin. http://dx.doi.org/10.1007/978-1-4612-3738-9
- Williams, C.H., et al. (1990) Malignant hyperthermia induction in susceptible swine following exposure to arduan. Anesthesia & Analgesia, 70, S433. http://dx.doi.org/10.1213/00000539-199002001-00433
- Williams, C.H. et al. (1985) Porcine malignant hyperthermia: Testing of atricurium in MH susceptible pigs. Anesthesia & Analgesia, 64, 112.
- Williams, C.H. and Hoech, G.P. (2008) Incidence of malignant hyperthermia in Greater Kansas City. #20.3. The integrative biology of exercise-V, 24-27 September.