Journal of Environmental Protection
Vol. 4 No. 9 (2013) , Article ID: 36316 , 8 pages DOI:10.4236/jep.2013.49107
Sorption of Denatonium to Kaolinite Clay from Water
![]()
Department of Chemistry and Biochemistry, University of Dayton, Dayton, USA.
Email: *gcrosson1@udayton.edu
Copyright © 2013 Garry S. Crosson et al. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Received January 11th, 2013; revised February 16th, 2013; accepted March 1st, 2013
Keywords: Denatonium; Sorption Isotherms; Kaolinite; Clay Minerals; Water Quality
ABSTRACT
Denatonium benzoate is an aversive agent proposed for use in certain anti-freeze formulations to reduce poisoning events of pets and humans. Unfortunately, the sorption properties of denatonium cations to soil and/or soil components have not been investigated. In this paper, denatonium sorption to reference kaolinite clay under varied pH, ionic strength, and cation identity is described. The data clearly indicate that the nature of the background electrolyte cation modulates denatonium sorption with enhanced sorption occurring the presence of potassium ions versus minimal sorption occurring in a calcium chloride electrolyte at each pH studied as estimated using Dubinin-Raduskevich (D-R) isotherms. The effect of background electrolyte concentration on sorption was consistent with an ion-exchange process wherein the sorption capacity increased with decreased background electrolyte concentration. An interesting and unexpected observation of increased sorption capacity (estimated using the D-R model) with decreasing pH was observed in 10 mM CaCl2 in contrast to an observed and expected increase in denatonium sorption to kaolinite in the presence of either of 10 mM NaCl or 10 mM KCl. Estimates of the mean sorption energy using D-R isotherms ranging from 2.65 kJ/mol in 10 mM CaCl2 at pH 4 to 4.68 kJ/mol at pH 10 suggesting that the sorption process is not exclusively ion-exchange.
1. Introduction
The proper stewardship of water supplies is essential to human health and quality of life. The use of any material which could negatively impact natural water supplies should thus be viewed with caution. Denatonium benzoate (DB), benzyldiethyl(2,6-xylylcarbamoylmethyl)ammonium benzoate is a water-soluble, quaternary ammonium compound (QAC) shown in Figure 1. DB is widely used to a) prevent human or animal consumption of poisonous substances because the compound is extremely bitter and b) to denature alcohol and prevent the sale and consumption of untaxed spirits [1-3]. Since 1995, Oregon has mandated that DB be added to products meeting certain criteria (windshield washer fluids containing > 4% methanol and automotive antifreeze containing > 10% ethylene glycol). More recently, The Antifreeze Bittering Act of 2005 (H.R. 2537) was reintroduced (H.R. 615, 2009) to amend the Federal Hazardous Substances Act and required engine coolant and antifreeze to contain a bittering agent (DB) to discourage ingestion by humans and pets. While well-spirited, the literature is devoid of studies addressing potential deleterious effects of DB and the environmental fate and mobility of DB, as highlighted in 2005 by Tom Bonacquisti, American Water Works Representative, in an address to the Environment and Hazardous Materials Subcommittee, of the House of Representatives Energy and Commerce Committee pertaining to The Antifreeze Bittering Act; in the address it was stated that “… little was known about the environmental fate and transport of DB...” and that “… small amounts of DB could render drinking water supplies bitter and unpalatable due to the extreme bitterness of DB” [4]. Since the literature lacks specific studies of DB partitioning between aqueous and solid phases, the sorptive behavior of related quaternary ammonium compounds can provide clues.
Quaternary ammonium compounds (QACs) are commonly utilized in consumer products, cleaning agents and industrial processes [5]. Benzalkonium chloride (BAC), for example, represents the most well-known member of the class and is used in antiseptic formulations in cosmetics, soaps, baby lotions, shampoos, eye drops and contact lense cleaning solutions; as a cleaning agent, BAC is utilized in hospitals to cleanse floors, surgical apparati and medical instruments [6,7]. Industrially, paper and pulp wastewater are treated with QAC-functionalized molecular sieves to remove dyes [4]; waters contaminated with pesticides (i.e. linuron) are cleaned via sorption to QAC treated wood [8] or sorption to QAC treated soils [9]. Anticholinergic drugs also utilize QACs as active ingredients [10]. Until recently, herbicides containing the toxic QACs, paraquat and diquat, are routinely applied to soils as herbicides [11]. Unfortunately, some have been linked to occupational asthma and other health problems [7].
Sediment and soil sorption of surfactant QACs is wellstudied [5,6,12,13]. Generally, surfactant results suggest that the quaternary ammonium cation adsorbs to mineral surfaces via a cation-exchange process with charge balancing cations associated with mineral surfaces. In clay minerals, such as 2:1 layered aluminosilicates, excess negative charge (due to isomorphic substitutions of cations into the clay structure) are present; these charges are generally balanced by alkali or alkaline earth cations [14]. QAC sorption to soil OM has also been investigated and indicates that herbicide sorption to montmorillonite is negatively impacted by organic matter [15].
In this preliminary study, the sorption properties of DB onto a reference kaolinite clay were investigated. The maximum sorbate concentration of DB was determined via sorption under varied solution conditions (i.e. pH, ionic strength, background cation identity).
2. Experimental
2.1. Materials
Denatonium benzoate was obtained from MP Biomedicals (USA). High-defect kaolinite (KGa-2), Warren County, Georgia was obtained from the Source Clay Minerals Repository at Purdue University and fractionated to obtain clay sized particles (<2 μm diamter). Burgos et al. reported a CEC value for KGa-2 43 at pH 7 in 10 mM LiCl [16]. All solvents were HPLC grade.
2.2. Batch Adsorption Experiments
Stock solutions of DB 2 g/L were prepared by DB dissolution in electrolytes matching those of the given ex-
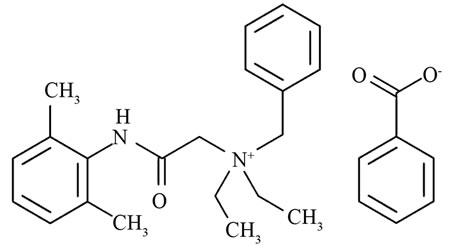
Figure 1. The structure of denatonium benzoate with the quaternary ammonium cation and the benzoate anion.
periment (i.e. 0.01 M CaCl2, 0.01 M NaCl, 0.01 M KCl, etc). Batch equilibrium adsorption experiments were implemented according to Organisation for Economic Cooperation and Development (OECD) guidelines [17]. The pH each of suspension was adjusted with dilute HCl or NaOH. All sorption experiments were performed in duplicate using Clarified Polypropylene, BD Falcon 50 mL conical centrifuge tubes. The reaction mixtures were equilibrated in a Big-shot III hybridization oven (Boekel Scientific) which mixed the samples end-over-end at 25 rpm for 24 hr. Equilibration studies indicated that 24 hrs was sufficient for attaining equilibrium in all systems. Subsequent to each experiment, all vials were centrifuged for 25 minutes at 4150 g. The pH of the supernatant was measured using a calibrated combination electrode.
2.3. Detection of Denatonium Benzoate
Aqueous denatonium concentrations were determined in two milliliter aliquots of the supernatant via HPLC (Perkin Elmer, Series 275 HRes UV/VIS Liquid Chromatography System with autosampler) using a C-18 analytical column (LiChrospher, 100 × 4.6, 5 μm). The mobile phase consisted of acetonitrile (ACN) and MilliQ water adjusted to pH 2.2 with H3PO4. The column flow rate was 1.0 mL·min−1. The total time analysis time for each sample was 59 minutes. The mobile phase composition was initially held constant at 70% water (solvent A) from 0 to 8 min, linearly ramped to 60% water from 8 to 14 min, held constant at 60% water from 14 to 23 min, linearly ramped to 30% water from 23 - 28 min, held constant at 30% water from 28 - 43 min and finally linearly ramped back to 70% water. The retention time of denatonium benzoate was 15.5 min in standards, but varied from 14.9 to 15.8 min in supernatant solutions suggesting that dissolved solutes from the clay mineral were present.
Denatonium was detected via absorption at 205 nm with 600 nm as the reference wavelength. Linearity up to 100 ppm DB in all electrolytes was consistently achieved with R2 = 0.999. Controls and blank experiments were run for each denatonium solution. Control samples consisted of denatonium benzoate solutions without clay minerals and experiment blank samples were composed of clay suspensions without denatonium benzoate. At the lowest denatonium concentration utilized, no changes in the aqueous concentration were detected in controls indicating that significant sorption by centrifuge walls were not likely. Thus, aqueous denatonium benzoate concentrations were determined by calculating the difference between the initial and final concentrations.
2.4. Sorption Isotherms
To predict or assess the potential for denatonium transport in subsoil environments, a number of empirical models are typically employed to estimate partition or distribution coefficients and to approximate the sorption capacity under a variety of solution conditions. The Freundlich isotherm is used to model the sorption capacity (qe, mmol/g) as a function of equilibrium solute concentration (Ce, mmol/L) allowing for heterogeneous surfaces and allow for multilayer sorption [18]. The Freundlich equation is given by
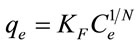 (1)
(1)
where KF, Ce and N represent the Freundlich factor, the equilibrium solute concentration, and surface heterogeneity, respectively. In contrast to the Freundlich sorption model, the Langmuir model is generally applicable for describing sorption to homogenous surfaces where monolayer absorption is presumed to occur. The Langmuir model is defined as
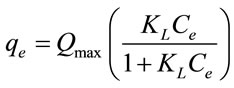 (2)
(2)
where Qmax is the maximum sorption capacity (mmol/g), KL is the Langmuir constant (L/g), and Ce is the equilibrium solute concentration (mmol/L).
To gain insight into potential sorption mechanisms, the Dubinin-Radushkevich (D-R) model can be utilized. The linearized form of the D-R model is [19]
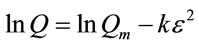 (3)
(3)
where Qm represents the Dubinin-Radushkevich monlayer capacity (mmol/g), k is a constant which allows the mean free energy of sorption to be calculated, and 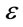 is the Polyani potential which can be related to the equilibrium concentration (Ce) as follows:
is the Polyani potential which can be related to the equilibrium concentration (Ce) as follows:
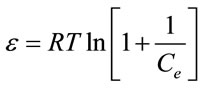 (4)
(4)
where R represents the molar gas constant 8.314 J·mol−1·K−1 and T is the absolute temperature. To determine the mean free energy change (E) in kJ/mol associated with a mole of molecules being transferred from solution to the sorbent surface, the following relationship can be utilized
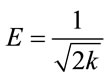 (5)
(5)
where k represents the slope of the Dubinin-Radushkevich model given in Equation (3).
3. Results and Discussion
3.1. pH Effects
Equilibrium sorption isotherms for denatonium sorption in CaCl2, KCl, and NaCl at pH 4.0, 6.5, and 10 shown in Figure 2. Generally, denatonium sorption to kaolinite in each electrolyte varies as a function of pH. Figure 2(a) shows the sorption behavior of denatonium in 0.01 M CaCl2 electrolyte at pH 4, 6.5, and 10 where the sorption isotherm is S-type [20] and sorption is inversely dependent on initial pH. A C-type [20] isotherm was observed in a potassium chloride electrolyte at all pH values (Figure 2(b)) along with a direct (albeit moderate) dependence of concentration on pH in contrast to the calcium chloride electrolyte. When sorption experiments were carried out in NaCl, L-type isotherms [20] were obtained under all solution conditions as shown in Figure 2(c). Generally, the Freundlich model satisfactorily fit all data with R2 > 0.91 as shown in Table 1. The suitableness of the Langmuir model considerably varied from system to system, and values derived from the model were not considered reliable (although they are shown in Table 1).
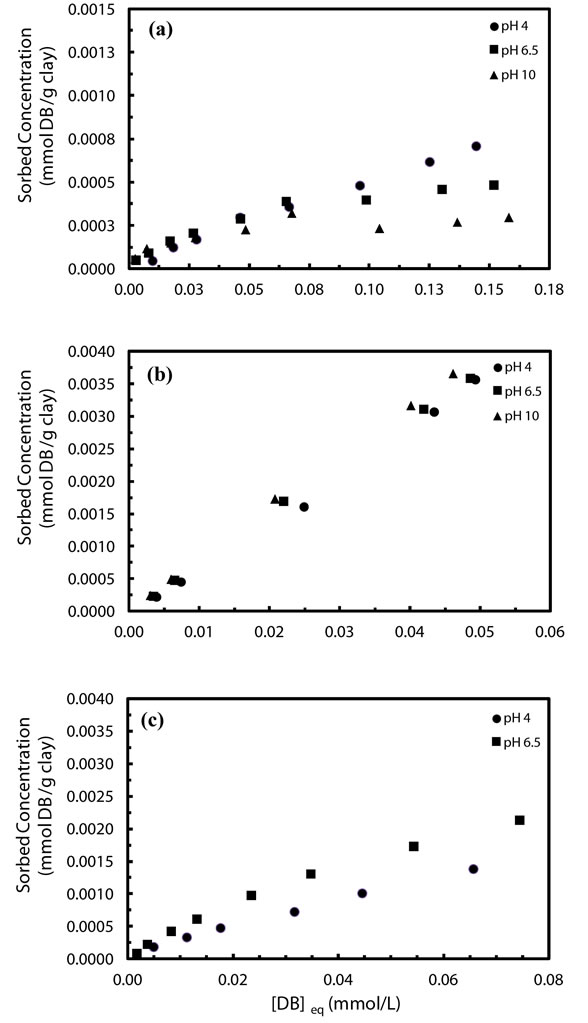
Figure 2. Sorption isotherms of denatonium by kaolinite in mM (a) Ca2+, (b) K+, and (c) Na+ at indicated pH values.
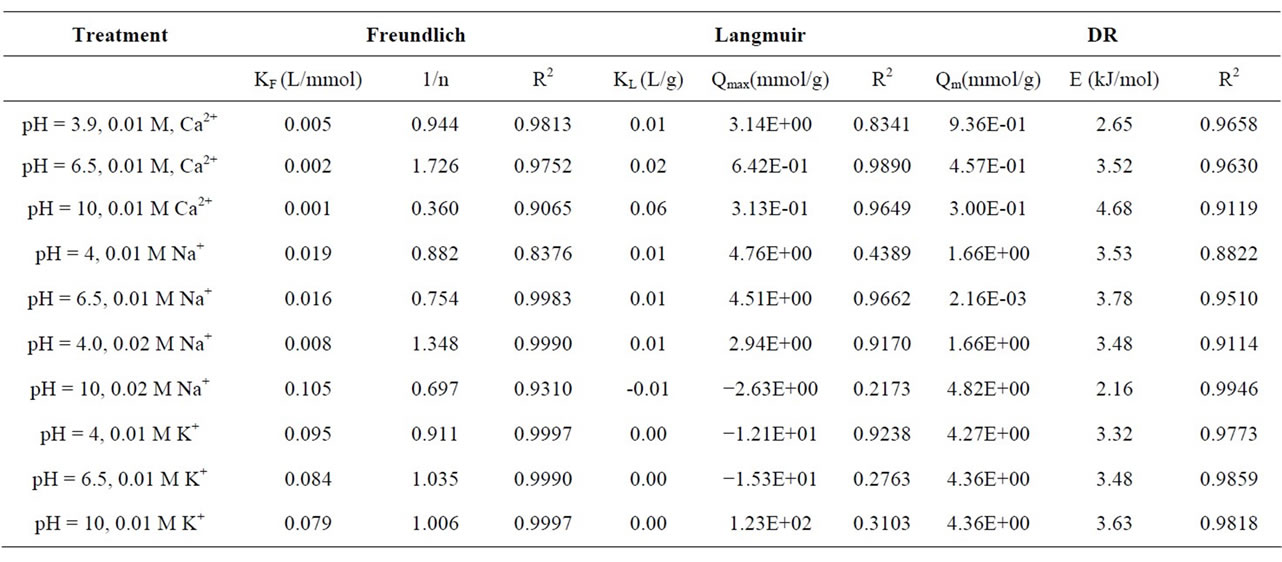
Table 1. Fitting parameters for Freundlich, Langmuir and Dubinin-Radushkevich (DR) isotherms.
Three principal observations can be made for denatonium sorption in 0.01 M CaCl2 over the pH range studied. Firstly, mineral sorption of denatonium varies with pH with the greatest amount of sorption occurring at pH 4 for the highest initial concentration. Secondly, at low initial denatonium concentrations (0 - 50 ppm), sorption is nearly independent of initial pH. Finally, the maximum sorption (at the highest initial concentration) varies inversely with pH with the lowest amount of sorption occurring at pH 10. However, in the presence of 0.01 M KCl, a positive correlation between initial pH and sorption was observed although the degree of sorption at different pH values was not large. A background electrolyte of 0.01 M NaCl yielded sorption isotherms which showed that denatonium sorption varied significantly with pH, in contrast to observations for 0.01 M KCl systems at the same pH. That the degree of sorption varies with initial pH is consistent with the fact that kaolinite has a pH-dependent surface charge due to protonation/deprotonation reactions of silanol and aluminol [14]. Generally, the net surface charge for kaolinite is expected to be zero at the point of zero charge (PZC) which has been found to be around 2.8 [21]. Thus, cation sorption is expected to occur and increase with initial solution pH for values in excess of the PZC. This has been observed in numerous studies of cation absorption to kaolinite [5,22,23]. For example, in an early study by Brownawell et al., the adsorption of the cationic surfactant dodecylpyridinium (DP) to kaolinite clay under varied solution conditions (pH, background electrolyte and dodecylpyridinium concentration) was found to be directly proportional to solution pH [5]. A similar observation was made in studies of paraquat [22] and methylene blue [23] dye sorption onto kaolinite.
Interestingly, the pH-dependent behavior observed for the 0.01 M CaCl2 system is counter to that which would be expected were a simple cation-exchange process occurring. To the contrary, as pH increased, the maximum amount of sorption generally decreased suggesting that the clay surface charge actually increased. A possible explanation could be that the background electrolyte (CaCl2) plays an active (although unknown) role in the sorption process wherein a partially dissociated calcium benzoate complex (CaBenzoate+) forms with the complex subsequently absorbing to the clay surface. The presence of such a complex was documented in an early study of organic calcium complexes where an association constant was measured in 5 mM CaCl2 and 50 mM sodium benzoate and found to be 0.75 suggesting that the complex has a small propensity to form [24,25]. Thus, although this complex was not readily detected in these experiments, it’s presence on the kaolinite surface could preclude the denatonium cation from sorbing to the surface and account for decreasing denatonium sorption at elevated pH values. An additional, though highly speculative, explanation could be that at pH 10, Ca(OH)2 precipitates could physically block access of denatonium cations to the surface thus reducing sorption. The obstructing of cation access to sorption sites in mineral systems was suggested in a study of copper sorption to layer silicates in natural soils; in the study Fe oxide precipitates were postulated to coat lateral surfaces of the silicates hindering the access of Cu to those sorption sites [26].
Denatonium sorption in the presence of 0.01 M KCl (Figure 2(b)) was markedly different from that occurring in the presence of calcium at the same concentration. The most notable difference is the linearity of sorption across the concentration range. Linear sorption isotherms suggest that under the given experimental conditions, binding at one site does not impact other binding sites of the sorbent such that the sorbate distribution constant does not vary with initial concentration and could further imply a solute partition mechanism rather than true sorption [27]. More likely than not, the linearity of isotherms observed here in the presence of potassium ions are more related to cation properties, i.e. low hydration energy, and the concentration range employed. Regardless, under the conditions of the study, it is apparent that the sorption process differs somewhat from that observed for sorption in the presence of CaCl2 and NaCl.
Figure 2(c) shows sorption isotherms collected in a 0.01 M NaCl background electrolyte at different pH values. Immediately apparent is a modulation in sorption mechanism in the concentration range by pH as evidenced by a change in isotherm shape. At low pH, the isotherm is linear across the range, however, with increasing pH, the isotherm displays Langmuir-type sorption again suggesting that monolayer sorption of denatonium to the kaolinite surface took place. The figure also shows that sorption increased with pH as would be expected were cation sorption to pH-dependent edge sites occurring [28].
3.2. Impact of Cation Concentration
To further assess the mobilization potential of denatonium in subsurface environments due to changes in solution composition, the impact of ionic strength on sorption in NaCl was evaluated. Figure 3 shows sorption isotherms collected at pH 4 in 0.01 and 0.02 M NaCl. The isotherms clearly indicate that cation concentration plays a role in the sorption process with higher concentrations of cation appearing to suppress sorption. Freundlich constants listed in Table 1 quantitatively support the observation of decreased affinity at increased ionic strength (0.019 L/mmol(0.01 M Na+)) vs. (0.008 L/mmol (0.02 M Na+)). The decrease in sorption observed at higher ionic strength can be rationalized by competition between denatonium cations and Na+ for sorption sites with denatonium cations likely forming outer sphere complexes in the background electrolyte. The observations suggest that sorption proceeds via a cation exchange mechanism. In previous studies of ion sorption to charged surfaces, the observation of reduced sorption in the presence of larger electrolyte concentrations was taken as evidence of an ion-exchange mechanism whereby sorption occurs via an outer-sphere complexation process [29,30]; our team thus proposes that outer-sphere complexation occurs between denatonium and the charged surfaces of kaolinite across the pH range.
3.3. Electrolyte Cation Identity Effects
To assess the impact of varying the electrolyte cation, we
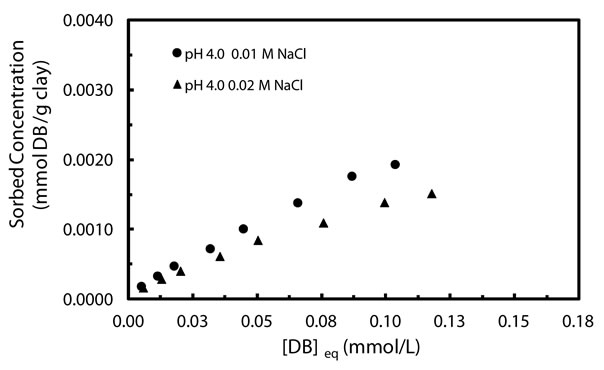
Figure 3. Sorption isothems of denatonium by kaolinite in 10 mM and 20 mM Na+ at pH 4.
evaluated sorption characteristics in the presence of Ca2+, K+, and Na+ at three initial pH values (4, 6.5 and 10) at varied denatonium concentrations up to 75 ppm. Figure 4 shows the sorption isotherms for each electrolyte at different pH values with all figures drawn to the same scale for comparison. Irrespective of initial pH, the affinity of denatonium for kaolinite decreases in the order K+ > Na+ > Ca2+. That denatonium sorption is enhanced in the presence of potassium ions and suppressed with calcium ions at all pH values is directly related to the hydration properties of potassium and calcium as previously suggested [31]. Ca2+ has an enthalpy of hydration of −1580 kJ/mol while that of K+ is −314 kJ/mol. The impact of saturating cations and supporting electrolyte cations on contaminant sorption to clay minerals has been well documented in the literature and our results are generally consistent with previous studies in that sorption is enhanced in the presence of low hydration enthalpy ions and suppressed in the presence of higher hydration energy, divalent cations [31- 33]. As observed in previous studies of contaminant sorption in the presence of the same cations (K+, Na+, Ca2+), clay retention of contaminants with sodium ions present in the background electrolyte was intermediate [32].
3.4. Sorption Mechanism
Representative D-R plots are shown for denatonium sorption to kaolinite in the presence of 0.01 M KCl at pH 4.0, 6.5, 10 are shown in Figure 5. With few exceptions, the linearized form of the Dubinin-Radushkevich model yielded satisfactory R2 values as shown in Table 1. The calculated mean free energy values ranged from +2.16 kJ/mol to +4.68 kJ/mol. In previous studies, many (but not all researchers) have a ascribed a magnitude of E between 8 and 16 kJ/mol to sorption processes proceeding via ion-adsorption or chemisorption [34-36] while values less than 8 kJ/mol are typically ascribed to
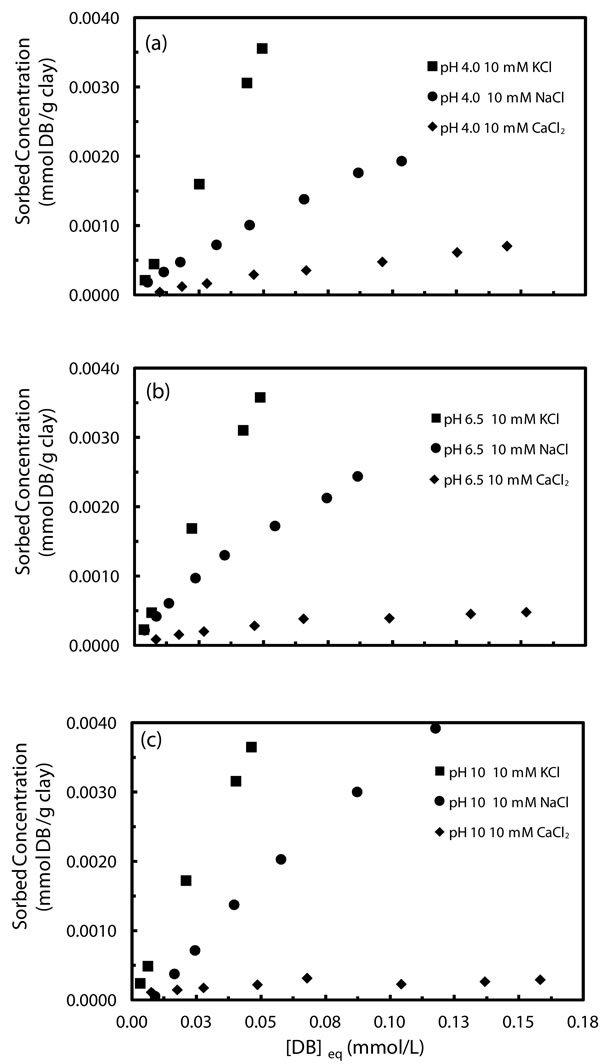
Figure 4. Sorption isotherms of denatonium by kaolinte in 10 mM electrolyte at (a) pH 4, (b) pH 6.5, and (c) pH 10.

Figure 5. Dubinin-Rudschevich isotherm for denatonium sorp tion onto kaolinite in 10 Mm KCI at different pH values.
physical sorption processes which tend to be rapid and reversible [37,38]. That our calculated values fall out of the range of ion-exchange sorption processes is in direct contrast to the experimentally observed sorption dependence on ionic strength as shown in Figure 3 for denatonium sorption in 0.01 M NaCl and 0.02 M NaCl. As discussed previously, this observation is generally indicative of a cation exchange mechanism being operative. Thus, the calculation of sorption energies outside the empirically accepted range for cation-exchange processes does not preclude a conclusion of cation exchange taking place; the observation further highlights the shortcomings of attempting to exclusively utilize theoretical models as the sole determinant of sorption mechanisms.
4. Conclusion
The sorption of the denatonium cation to specimen clay samples has been evaluated under a variety of solution conditions. Kaolinite sorption of denatonium is very much influenced by pH, ionic strength, and by electrolyte cation identity. At low initial concentrations of denatonium, equilibrium denatonium concentrations were independent of pH for all electrolytes studied but varied at higher initial concentrations. The uptake of denatonium was maximal at low initial pH values in CaCl2 while in KCl and NaCl maximum sorption was observed at higher pH values. An observed inverse dependent sorption with ionic strength was taken as evidence of a cation exchange mechanism being operative in each case.
5. Acknowledgements
This research was funded, in part, by the United States Geological Survey, State Water Resources Research Institute Program of Ohio through grant 2010OH169B. Finally, Department of Chemistry and Biochemistry and University of Dayton Research Institute provided support for this work through faculty start-up funds provided to GSC.
REFERENCES
- M. C. Henderson, C. M. Neumann and D. R. Buhler, “Analysis of Denatonium Benzoate in Oregon Consumer Products by HPLC,” Chemosphere, Vol. 36, No 1, 1998, pp. 203-210. doi:10.1016/S0045-6535(97)10033-9
- D. Zuba, C. Oewiegoda, B. Byrska and W. Lechowicz, “Assessment of the Effectiveness of Denationium Benzoate (BITREX) Removal from Denatured Spirit Preparations Using Sodium Hypochlorite,” Problems of Forensic Sciences, Vol. 113, 2005, pp. 288-298.
- D. Zuba, C. Oewiegoda, B. Byrska and W. Lechowicz, “Determination of Denatonium Benzoate (BITREX) in Denatured Spirit Preparations,” Problems of Forensic Sciences, Vol. 113, No. 2005, pp. 275-287.
- US House of Representatives, “Statement of the American Waterworks Association before the Energy and Hazardous Materials Subcommittee Concerning H.R. 2537 The Antifreeze Bittering Act of 2005,” Energy and Hazardous Materials Subcommittee Committee of Energy and Commerce, 2005.
- B. J. Brownawell, H. Chen, J. M. Collier and J. C. Westall, “Adsorption of Organic Cations to Natural Materials,” Environmental Science & Technology, Vol. 24, No. 8, 1990, pp. 1234-1241. doi:10.1021/es00078a011
- J. A. Smith, P. R. Jaffe and C. T. Chiou, “Effect of Ten Quaternary Ammonium Cations on Tetrachloromethane Sorption to Clay from Water,” Environmental Science & Technology, Vol. 24, No. 8, 1990, pp. 1167-1172. doi:10.1021/es00078a003
- A. Purohit, M. C. Kopferschmitt-Kubler, C. Moreau, E. Popin, M. Blaumeiser and G. Pauli, “Quaternary Ammonium Compounds and Occupational Asthma,” International Archives of Occupational and Environmental Health, Vol. 73, No. 6, 2000, pp. 423-427. doi:10.1007/s004200000162
- M. S. Rodriguez-Cruz, M. S. Andrades, A. M. Parada, M. J. M. Sanchez-Martín, “Effect of Different Wood Pretreatments on the Sorption-Desorption of Linuron and Metalaxyl by Woods,” Journal of Agricultural and Food Chemistry, Vol. 56, No. 16, 2008, pp. 7339-7346. doi:10.1021/jf800980w
- M. Mingorance, L. Sanchez and A. Pena, “Pesticide Adsorption on a Calcareous Soil Modified with Sewage Sludge and Quaternary Alkyl-Ammonium Cationic Surfactants,” Environmental Science and Pollution Research, Vol. 15, No. 1, 2008, pp. 8-14. doi:10.1065/espr2007.02.387
- M. M. Ariffin and R. A. Anderson, “LC/MS/MS Analysis of Quaternary Ammonium Drugs and Herbicides in Whole Blood,” 43rd International Conference of the International-Association-of-Forensic-Toxicologists (TIAFT), Seoul, 29 August-2 September 2005, pp. 91-97.
- M. Pateiro-Moure, E. Martinez-Carballo, M. Arias-Estevez and J. Simal-Gandara,”Determination of Quaternary Ammonium Herbicides in Soils-Comparison of Digestion, Shaking and Microwave-Assisted Extractions,” 10th International Symposium on Advances in Extraction Techniques, Bruges, 28-30 January 2008, pp. 110-116.
- C. Willms, Z. H. Li, L. Allen and C. V. Evans, “Desorption of Cesium from Kaolinite and Illite Using Alkylammonium Salts,” Applied Clay Science, Vol. 25, No. 3-4, 2004, pp. 125-133. doi:10.1016/j.clay.2003.10.001
- S. A. Boyd, M. M. Mortland and C. T. Chiou, “Sorption Characteristics of Organic-Compounds on Hexadecyltrimethylammonium-Smectite,” Soil Science Society of America Journal, Vol. 52, No. 3, 1988, pp. 652-657. doi:10.2136/sssaj1988.03615995005200030010x
- D. L. Sparks, “Environmental Soil Chemistry,” 4th Edition, Academic Press, San Diego, 1995.
- M. Pateiro-Moure, C. Perez-Novo, M. Arias-Estevez, R. Rial-Otero and J. Simal-Gandara, “Effect of Organic Matter and Iron Oxides on Quaternary Herbicide sorptionDesorption in Vineyard-Devoted Soils,” Journal of Colloid and Interface Science, Vol. 333, No. 2, 2009, pp. 431-438. doi:10.1016/j.jcis.2009.02.019
- W. D. Burgos, N. Pisutpaisal, M. C. Mazzarese and J. Chorover, “Adsorption of Quinoline to Kaolinite and Montmorillonite,” Environmental Engineering Science, Vol. 19, No. 2, 2002, pp. 59-68. doi:10.1089/10928750252953697
- OECD, “Test No. 106: Adsorption: Desorption Using a Batch Equilibrium Method,” OECD. Paris, 2000. doi:10.1787/9789264069602-en
- R.-L. Tseng and F.-C. Wu, “Inferring the Favorable Adsorption Level and the Concurrent Multi-Stage Process with the Freundlich Constant,” Journal of Hazardous Materials, Vol. 155, No. 1-2, 2008, pp. 277-287. doi:10.1016/j.jhazmat.2007.11.061
- T. Shahwan and H. N. Erten, “Thermodynamic Parameters of Cs+ Sorption on Natural Clays,” Journal of Radioanalytical and Nuclear Chemistry, Vol. 253, No. 1, 2002, pp. 115-120. doi:10.1023/A:1015824819940
- G. Limousin, J. P. Gaudet, L. Charlet, S. Szenknect, V. Barthes and M. Krimissa, “Sorption Isotherms: A Review on Physical Bases, Modeling and Measurement,” Applied Geochemistry, Vol. 22, No. 2, 2007, pp. 249-275. doi:10.1016/j.apgeochem.2006.09.010
- C. Appel, L. Q. Ma, R. Dean Rhue and E. Kennelley, “Point of Zero Charge Determination in Soils and Minerals via Traditional Methods and Detection of Electroacoustic Mobility,” Geoderma, Vol. 113, No. 1-2, 2003, pp. 77-93. doi:10.1016/S0016-7061(02)00316-6
- K. Draoui, R. Denoyel, M. Chgoura and J. Rouquerol, “Adsorption of Paraquat on Minerals. A Thermodynamic Study,” Journal of Thermal Analysis and Calorimetry, Vol. 58, No. 3, 1999, pp. 597-606. doi:10.1023/A:1010152411288
- A. R. Tehrani-Bagha, H. Nikkar, N. M. Mahmoodi, M. Markazi and F. M. Menger, “The Sorption of Cationic Dyes onto Kaolin: Kinetic, Isotherm and Thermodynamic Studies,” Desalination, Vol. 266, No. 1-3, 2011, pp. 274- 280. doi:10.1016/j.desal.2010.08.036
- N. R. Joseph, “The Dissociation Constants of Organic Calcium Complexes,” Journal of Biological Chemistry, Vol. 164, No. 2, 1946, pp. 529-541.
- S. Sen, P. Yu, V. P. Klyuev and B. Z. Pevzner, “Atomic Structure of Beryllium Boroaluminate Glasses: A MultiNuclear NMR Spectroscopic Study,” Journal of NonCrystalline Solids, Vol. 354, No. 33, 2008, pp. 4005-4011. doi:10.1016/j.jnoncrysol.2008.06.002
- A. G. Christy, “A Monte Carlo Study of Shortand LongRange Order of Tetrahedral Cations in Sapphirine and Khmaralite,” American Mineralogist, Vol. 94, No. 2-3, 2009, pp. 270-282. doi:10.2138/am.2009.2956
- P. Behra, E. Lecarme-Theobald, M. Bueno and J. J. Herhardt, “Sorption of Tributyltin onto a Natural Quartz Sand,” Journal of Colloid and Interface Science, Vol. 263, No. 1, 2003, pp. 4-12. doi:10.1016/S0021-9797(03)00238-8
- D. A. Ellis, J. W. Martin, D. C. G. Muir and S. A. Mabury, “Development of an 19F NMR Method for the Analysis of Fluorinated Acids in Environmental Water Samples,” Analytical Chemistry, Vol. 72, No. 4, 2000, pp. 726-731. doi:10.1021/ac9910280
- W. Yan, S. Hu and C. Jing, “Enrofloxacin Sorption on Smectite Clays: Effects of pH, Cations, and Humic Acid,” Journal of Colloid and Interface Science, Vol. 372, No. 1, 2012, pp. 141-147. doi:10.1016/j.jcis.2012.01.016
- H. Gulley-Stahl, P. A. Hogan, W. L. Schmidt, S. J. Wall, A. Buhrlage and H. A. Bullen, “Surface Complexation of Catechol to Metal Oxides: An ATR-FTIR, Adsorption, and Dissolution Study,” Environmental Science & Technology, Vol. 44, No. 11, 2010, pp. 4116-4121. doi:10.1021/es902040u
- H. Li, B. J. Teppen, D. A. Laird, C. T. Johnston and S. A. Boyd, “Geochemical Modulation of Pesticide Sorption on Smectite Clay,” Environmental Science & Technology, Vol. 38, No. 20, 2004, pp. 5393-5399. doi:10.1021/es0494555
- S. B. Haderlein and R. P. Schwarzenbach, “Adsorption of Substituted Nitrobenzenes and Nitrophenols to Mineral Surfaces,” Environmental Science & Technology, Vol. 27, No. 2, 1993, pp. 316-326. doi:10.1021/es00039a012
- G. Y. Sheng, C. T. Johnston, B. J. Teppen and S. A. Boyd, “Adsorption of Dinitrophenol Herbicides from Water by Montmorillonites,” Clays and Clay Minerals, Vol. 50, No. 1, 2002, pp. 25-34. doi:10.1346/000986002761002630
- K. Gupta, T. Basu and U. C. Ghosh, “Sorption Characteristics of Arsenic(V) for Removal from Water Using Agglomerated Nanostructure Iron(III)-Zirconium(IV) Bimetal Mixed Oxide”, Journal of Chemical & Engineering Data, Vol. 54, No. 8, 2009, pp. 2222-2228. doi:10.1021/je900282m
- L. Mihaly Cozmuta, A. Mihaly Cozmuta, A. Peter, C. Nicula, E. Bakatula Nsimba and H. Tutu, “The Influence of pH on the Adsorption of Lead by Na-Clinoptilolite: Kinetic and Equilibrium Studies,” Water Sa, Vol. 38, No. 2, 2012, pp. 269-278.
- Y. S. Ho, J. F. Porter and G. McKay, “Equilibrium Isotherm Studies for the Sorption of Divalent Metal Ions onto Peat: Copper, Nickel and Lead Single Component Systems,” Water, Air and Soil Pollution, Vol. 141, 2002, pp. 1-33.
- S. S. Tahir and N. Rauf, “Removal of a Cationic Dye from Aqueous Solutions by Adsorption onto Bentonite Clay,” Chemosphere, Vol. 63, No. 11, 2006, pp. 1842- 1848. doi:10.1016/j.chemosphere.2005.10.033
- M. Akçay, “Characterization and Adsorption Properties of Tetrabutylammonium Montmorillonite (TBAM) Clay: Thermodynamic and Kinetic Calculations,” Journal of Colloid and Interface Science, Vol. 296, No. 1, 2006, pp. 16-21. doi:10.1016/j.jcis.2005.08.031
NOTES
*Corresponding author.