Journal of Environmental Protection
Vol. 3 No. 5 (2012) , Article ID: 19514 , 6 pages DOI:10.4236/jep.2012.35051
Oxidative Stress and Oxidative Damage in Male Rat Erythrocytes Associated with Prolonged Exposure to Smoke Pollution
![]()
Institute of Ecology and Environmental Studies, Obafemi Awolowo University, Ile-Ife, Nigeria.
Email: *aachudum@yahoo.com
Received February 1st, 2012; revised February 29th, 2012; accepted April 2nd, 2012
Keywords: Lipoperoxidation; Antioxidants; Oxidative Stress; Antioxidases; Smoke Pollution; Oxidative Damage; Erythrocytes
ABSTRACT
Exposure of animals to different duration of smoke can result in a wide range of adverse clinical conditions. Toxicological consequences arising from prolonged exposure may not be totally enumerated. The present study was to evaluate the oxidative stress-inducing potential of a short and long-time exposure of male rat erythrocytes to environmentally relevant to public health. Male Wistar rats were exposed to open municipal land-fill smoke for 30 and/or 60 days. The oxidative stress in erythrocytes was evaluated by assessing the magnitude of malondialdehyde production and reduced glutathione (GSH) contents and the activities of superoxide dismutase (SOD), catalase (C) and glutathione reductase (GR) after 30 and/or 60 days of exposure. After 30 days there was no change in SOD, catalase and GR activities and no alterations in GSH and lipid peroxidation (LPO). After 60 days, the levels and activities of SOD (23.62%), catalase (5.24%), GR (14.87%) decreased. Overall, the study demonstrates that the chronic exposure of male rats to smoke results in induction of oxidative stress and simultaneous reduction in antioxidative defense system in erythrocytes at 60 days of exposure.
1. Introduction
Many developed countries are confronted with the burden of air pollution and fire outrages on their forest reserves [1]. Globally, indoor air pollution from burning of biomass in smoky, inefficient stoves lead to nearly three million premature deaths each year [2]. Considerable air pollution occurs in big cities worldwide [3]. In the developing world, rapid population growth has translated into large urbanization with attendant increases in the volume of air pollution problems. Increases in vehicular exhaust coupled with industrial air pollution and workyard smoke continue to pollute the atmosphere unabated. Welding fumes also permeate the environment and open air burning from municipal waste and landfills are major contributors to air pollution problems [4]. Air pollution generally has a great impact on both local and global environment [5,6].
Smoke pollution assumes a great concern globally due to manifestation of technological and scientific innovations in various fields in addition to diverse activities of man for his sophistication. Recently, urban and non-urban pollution problems are developing in various activities namely, residential [7], agricultural [8,9], and small scale industrial activities [10]. This characteristics display a pronounced variability in time and space due to wide variety of information on particulate matter [11]. New information suggests that pollutants in smoke are more toxic than previously thought [7] because particle pollution is a silent killer [10].
Many studies have reported on the atmospheric pollutants particles during assessment of the environmental status. This is generally required to identify hazards that are present and to implement control. Follow up studies reported on the atmospheric particle size distribution and chemical nature by urban and rural influenced aerosols in many areas of the world [8]. Long and short-term exposure to low levels of particulate smoke pollution has shown to increase the risk of stroke [12], acute chronic bronchitis [13] and heart disease [14].
The decomposition products found in smoke and its fume such as oxides of nitrogen, phosgene, carbon monoxide and carbon dioxide in the environment necessitate this study. Data generated from this cohort will be used to assess the long-term effects of smoke pollution on lipoperoxidation, potential oxidative stress and how these might be affected by changes in exposure to smoke. The main aim of the present study is to report on the baseline data of smoke pollution on superoxide dismutase (SOD), catalase (C), glutathione reductase (GR) activities, glutathione and lipid peroxidation (LPO) levels in wistar rats.
2. Materials and Methods
The study area is geographically located at latitude 6˚10' N and 6˚15'N and longitude 2˚15'E and 4˚10'E. During December and January, predominant wind spread in NW, W and SW direction. The hazy Harmattan conditions were prevailing about 21.9% and predominant wind speed of 1 - 5 km/ph. Based on meteorological factors, synoptic scale wind pattern, topography, proper crosswind distribution in downwind direction, only four locations were selected. The location sites A (sham) lies between east and north-east side, the other two locations B and C lie between south-west and west direction. The control locations lie 2 km away from Municipal Refuse landfill.
2.1. Animals
Wistar rats, 8 - 10 wks, weighing 195 ± 10 g were procured from the animal house, Department of Anatomy and Cell Biology, Obafemi Awolowo University, Ile-Ife, Nigeria and acclimatized to the laboratory. Animals (40) males were randomized into 4 groups. Group A was placed in abandoned site A (sham) surrounded by commercial and traffic environment, whereas duplicate groups B and C were placed in the center of the smoke polluted area attributed by chunk car parts, mechanic workshops and commercial activities. The control location site lies 2 km from the open burning refuse landfill. The animals were housed in plastic cages (41 × 28 × 15 cm) under laboratory conditions. Rats were fed food pellets (Ladokun Feeds, Nigeria LTD) and water was provided ad libitum. Five animals were placed in each of the 8 boxes. Animal treatments and protocol employed in this study was according to the Institutional Ethical Committee, also the principles and guidelines as contained in the “Principles of laboratory Animal Care” (NIH publication No. 85 - 23) were followed.
2.2. Analytical Procedure
During the course of the experiment, two rats died in B and C groups, including one control; however, data of three animals in each group have been presented to maintain uniformity of sample size of all groups. None of the animals that died showed any visible signs of toxicity. At test termination, blood was collected from retro-orbital plexus with the help of capillary tube 24 h after 30 and/or 60 days of exposure. Heparin (2 mg/ml) was used as an anticoagulant. Blood samples were centrifuged at 2000 rpm for 15 min to separate plasma. The layer of white blood cells above the packed erythrocytes was discarded. Erythrocyte pellet was washed three times with 0.15 M NaCl, diluted (33%) in phosphate buffer saline (mM: NaCl, 136.9; KCl, 2.68; KH2·PO4, 1.47; and Na2·HPO4, 6.62; pH 7.4) and kept at 4˚C until further analysis. Liver was disserted out, ground and centrifuged, the homogenate and the 33% packed erythrocytes were used for the estimation of LPO, GSH, Glutathione reductase, Catalase and Superoxide dismutase. LPO and GSH were measured on the day of blood collection.
2.3. Measurement of LPO
LPO was assessed by measuring malondialdehyde (MDA) levels by thiobarbituric acid (TBA) reaction [15]. In brief, 1 ml of 33% packed erythrocytes was incubated at 37˚C ± 0.5˚C for 2 h. The sample was mixed with 1 ml of 10% w/v trichloroacetic acid to precipitate protein. The mixture was centrifuged at 2000 rpm for 10 min and an aliquot of 1 ml supernatant was reacted with 1 ml of 0.67% thiobarbituric acid in boiling water bath for 10 min. After cooling, it was diluted with 1 ml of distilled water and the absorbance was read at 535 nm. Results have been expressed as nmol/ml packed erythrocytes.
2.4. Assay of GSH Level
GSH content in erythrocytes was estimated by the method of Prins and Loos [16]. Briefly, an erythrocyte pack of 0.2 ml (33%) was mixed with 4 ml of 0.08 N H2SO4. After 10 min, 0.5 ml of tungstate solution was added and mixed vigorously for 5 min to clear the brown hemolysate. After allowing it to stand for 5 min to avoid crust formation on top of the supernatant, it was centrifuged for 15 min at 2000 rpm at room temperature. 2 milliliters of supernatant was mixed with 2.5 ml of Tris buffer (pH 8.0) and 0.2 ml of 5,5’-dithiobis-2-nitrobenzoic acid reagent. Within a minute, absorbance was read at 412 nm. The levels of GSH have been expressed as mmol GSH/ml packed erythrocytes.
2.5. Determination of SOD Activity
SOD activity was determined by the procedure of Madesh and Balasubramanian [17]. The reaction mixture contained 0.65 ml PBS (pH 7.4), 30 µl 3-(4-5 dimethyl thiazol 2-xl) 2,5-diphenyl tetrazolium bromide (MTT; 1.25 mm), 75 µl pyrogallol (100 µM), and 10 µl hemolysate (10%). The mixture was incubated at room temperature for 5 min, and the reaction was stopped by adding 0.75 ml of dimethyl sulfoxide. The absorbance was read at 570 nm and the activity has been expressed as Unit. One unit of SOD was defined as the micrograms of hemoglobin causing 50% inhibition in the MTT reduction.
2.6. Determination of Catalase Activity
Catalase activity in erythrocytes was assayed by the spectrophotometric method of Aebi [18]. Two ml of phosphate buffer (50 mM, pH 7.0) and 10 µl hemolysate (10%) were taken in a curvette. Reaction was started by adding 1 ml H2O2 (10 mM) and the absorbance was recorded at every 10 sec for 1 min at 240 nm against water blank. The activity of catalase has been expressed as mmol H2O2 utilized/min/mg hemoglobin.
2.7. Determination of GR Activity
GR activity was measured following the method of Goldberg and Spooner [19]. The 3 ml of reaction mixture contained 2.6 ml PBS 12 M, pH 7.2), 0.1 ml EDTA (15 mM), 0.1 ml oxidized glutathione (GSSG) (65.3 mM). To this, 10 µl of hemolysate was added and the volume was made up to 2.95 ml with distilled water. After incubation at room temperature for 5 min, 0.05 ml of NADPH (9.6 mM) was added. Decrease in absorbance/min was recorded immediately at 340 nm for 3 min. Control was run without GSSG. The activity of GR has been expressed as unit/g hemoglobin. One unit is µmol NADPH utilized/min/g hemoglobin.
Estimation of Hemoglobin in erythrocytes was determined by using kits (BDH Chemical, England) following the manufacturer’s recommendations.
2.8. Statistical Analysis
All values have been expressed as mean ± SEM. Two-way analysis of variance followed by Duncan’s post-hoc test was used to find out the differences between mean values. The differences were considered significant when p < 0.05.
3. Results
Effects of exposure to different durations to smoke on various oxidative stresses related parameters in rat erythrocytes are presented in Table 1. Results presented demonstrate that exposure to smoke for 30 days do not affect the alterations of the oxidative stress-related endpoints in rat erythrocytes. However, exposure to smoke for 60 days cause time-dependent effects on the different attributes evaluated for assessing the oxidative stressinducing potential of smoke in rat erythrocytes.
After 30 days of exposure, smoke neither increased nor altered the antioxidative parameters of rat erythrocytes except the activity of SOD which was 0.21% but not significant in comparison to control activity. Rats exposed in the center of smoke for 60 days increased LPO level by 2.05% of the control. Similarly, the effects are marked for GSH which increased by 2.38% and 4.76% for 30 and 60 days respectively. In the liver, smoke could not produce any significant changes in the magnitude of LPO and GSH levels respectively, as observed in the activities of catalase (0.73%), SOD (1.15%), GR (0.07%) when rats are exposed for 30 days. However, after 60 days of exposure, smoke cause significant increased in LPO (4.52%) p < 0.05, diminution in the activities of catalase (5.24%), SOD (23.62%), GR (14.87%), and GSH (19.15%) level.
4. Discussion
The present study shows that exposure to smoke for 30 days did not cause oxidative stress in rat erythrocytes. Although, short time exposure did not cause any increase
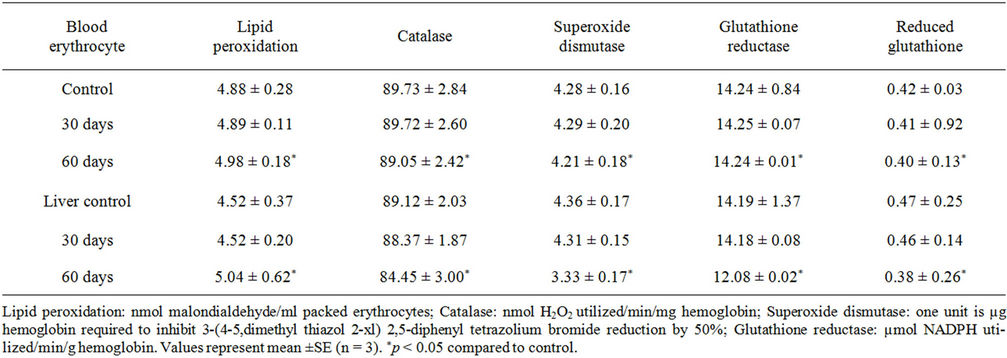
Table 1. Lipid peroxidation and antioxidative systems in erythrocytes of male rats exposed to smoke for 30 days and/or 60 days.
in LPO levels and showed no stress in antioxidative defense systems. Prolong exposure to smoke induced oxidative stress in rat erythrocytes (Table 1). After 30 days of exposure, smoke did not enhance LPO, and did not increase the levels of enzymatic and nonenzymatic antioxidants. This could be a compensatory system to combat the low-level reactive oxygen species (ROS) generation by smoke in the early phase of exposure and indicates that the development of oxidative stress is commiserating with the advancement of exposure duration.
Erythrocytes are more vulnerable to LPO [20]. Lipid peroxidation has been shown to cause profound alterations in the structure and functions of the cell membrane including decreased membrane fluidity, increased membrane permeability, inactivation of membrane-bound enzymes, and loss of essential fatty acids [21]. Several studies in occupationally exposed human subjects indicate that smoke enhance LPO and alter antioxidant systems in erythrocytes of human cutaneous model [22] and in electric arc welding operators [4,23]. The observed increase in 60 days exposures in the present study may be attributed to increased oxidative stress caused by the smoke-induced generation of ROS and depletion of erythrocyte sulfhydryl content coupled with decrease in enzymatic antioxidative defense mechanism, as reported by [4]. Reduction in enzyme activities with longer duration may be related to consumption of enzymes against oxidative stress. The decreased activity of the antioxidases could additionally be attributed to their nature of synergistic functioning, which may partly explain the mechanism of their reduced activity [24]; when catalase fails to eliminate H2O2 from the cell, the accumulated H2O2 causes inactivation of SOD [25].
Liver is the principal detoxification organ and the potential impact of smoke on all of the antioxidative attributes was almost similar to that of erythrocytes. The finding in this study showed that there existed in all likelihood, an imbalance between oxidation and antioxidation, and an oxidative stress and a potential oxidative damage in rats exposed to smoke. The principal mechanisms by which xenobiotics decrease the level of GSH are conjugation of GSH synthesis. In the present study, the percent changes in GR activity and GSH level were decreased by 0.07% and 2.13% after 30 days whereas, after 60 days by 14.87% and 19.15% of the control respectively. Thus, the reduced GR activity may not be the sole cause for depletion of GSH. Current findings indicate that GSH depletion may be related to oxidative reaction with the xenobiotics. In the present study, smoke pollution may have generated a large amount of superoxide anion radicals ( ), hydroxyl radicals (*OH), and singlet oxygen (1O2) [4], while, hydrogen peroxide H2O2 and other reactive species were similarly observed by [26]. Therefore, the depletion of erythrocyte GSH may also partly be related to the decreased SOD synthesis by decreased diminution of
), hydroxyl radicals (*OH), and singlet oxygen (1O2) [4], while, hydrogen peroxide H2O2 and other reactive species were similarly observed by [26]. Therefore, the depletion of erythrocyte GSH may also partly be related to the decreased SOD synthesis by decreased diminution of . Again, the GSH is the most sensitive antioxidant which protects the erythrocytes from the insult, either through the glutathione cycle or the mercapturic acid pathway [5]. Thus, it is assumed that toxic chemicals present in the smoke increases LPO thereby giving stress to the glutathione cycle. Reduced glutathione is a widely distributed cellular reductant and is a metabolic regulator and regular indicator of health.
. Again, the GSH is the most sensitive antioxidant which protects the erythrocytes from the insult, either through the glutathione cycle or the mercapturic acid pathway [5]. Thus, it is assumed that toxic chemicals present in the smoke increases LPO thereby giving stress to the glutathione cycle. Reduced glutathione is a widely distributed cellular reductant and is a metabolic regulator and regular indicator of health.
Taking together, this results coupled with existing environmental health problems in both developing and developed nations, is associated with numerous diseases [27]. Several diseases are due to an imbalance between the activities of an oxidant agent and antioxidant system within the cell. Toxicity associated with smoke could be due to oxidative tissue damage as they catalyze oxidative reactions in biological macromolecules. For example, International Agency for Research on cancer [28], reviewed the global evidence and classified household biomass of fuel smoke as a probable human carcinogen, while coal smoke was classified as a proven human carcinogen [28]. This interpreted to mean that biomass fuel is evidenced from fuel wood smoke [29], while, a longterm exposure to air pollution culminates in incidences of cardiovascular events in women [30]. The diversity of ideas about studies of outdoor air pollution and active and passive tobacco smoke show that heart disease could be expected from biomass indoor [28]. In addition, asthma might be expected as an outcome of fuel smoke. Nevertheless, studies have implicated biomass smoke to childhood pneumonia to having heart and lung effects in women [31,32].
In conclusion, the present study of potential toxicity associated with smoke demonstrates induction of oxidative stress in erythrocytes and liver of rat exposed for prolong duration, source of other numerous organ damage. Smoke induced depletion of GSH level is directly associated with elevation in lipid peroxidation which could be attributed to its protection against ROS generated by smoke. Therefore, smoke pollution induced oxidative stress smoke pollution induced oxidative stress and is associated with simultaneous reduction in erythrocyte and liver antioxidant defense systems. The relationship between the levels of exposure to smoke in the current study and the levels to which the general populations living in and around smoky environment are exposed is relevant to public health.
REFERENCES
- S. Leahy, “A Dozen Countries Take on Toxic Pollution,” North America Inter Press Service, Chicago, 2010.
- Blacksmith Institute, “The World’s Worst Polluted Places,” 2007. http://www.blacksmithinstitute.org/docs/costEff1.pdf
- WHO, “Public Health and Environment,” 2007.
- J. F. Zhou, D. Cai and G. Z. Tong, “Oxidative Stress and Potential Free Radical Damage Associated with Photocopying, a Role for Ozone?” Free Radical Research, Vol. 37, 2003, pp. 137-143. doi:10.1080/1071576021000036623
- V. K. Singh, M. K. Pathak, V. Bihari, Jyoti, D. K. Patel, N. Mathur, C. N. Kesavachandra and M. K. J. Siddiqui, “Studies on Oxidative Stress Induced Nerve Conduction Deficits in Cigarette Smokers,” Journal of Environmental Biology, Vol. 32, No. 1, 2011, pp. 39-42.
- A. C. Achudume, F. Aina and B. Onibere, “Pollution Tolerance of Smoke in the Distribution of Neuro-Transmitter Enzyme (Acetylcholine Esterase and High Density Cholesterol,” Journal Environmental Protection, Vol. 1, 2010, pp. 475-479. doi:10.4236/jep.2010.14055
- P. Wilkson, K. Smith, M. Joffe and A. Haines, “A Global Perspective on Energy: Health Effects and Injuries,” The Lancet, Vol. 370, No. 9591, 2007, pp. 965-978. doi:10.1016/S0140-6736(07)61252-5
- Y. C. Chan, P. D. Vowles, G. H. McTanish, R. W. Simpson, D. D. Cohen, G. M. Bailey and G. D. McOrist, “Characterization and Source Identification of PM-10 Aerosols Samples Collected with a High Volume Cascade Impactor in Brisbane (Australia),” Science of Total Environment, Vol. 262, No. 1-2, 2000, pp. 5-19. doi:10.1016/S0048-9697(00)00571-4
- P. R. Salve, R. J. Krupadam and S. R. Wate, “A Study on Major Inorganic Ion Composition of Atmospheric Aerosols,” Journal Environmental Biology, Vol. 28, No. 21, 2007, pp. 241-244.
- O. Taiwo, “The State of Urban Air Pollution in Lagos: Crisis of Management,” Lagos Metropolitant Area Transport Authority (LAMATA) Bulletin, 2005.
- S. Mullyta and K. Kuvaja, “Societal Premises for Sustainable Development in Large Southern Cities,” Global Environmental Change, Vol. 15, No. 3, 2005, pp. 224- 237. doi:10.1016/j.gloenvcha.2005.01.001
- D. G. Cook and D. P. Strachan, “Parental Smoking and Prevalence of Respiratory Symptoms and Asthma in School Age Children,” Thorax, Vol. 52, No. 12, 1997, pp. 1081- 1094. doi:10.1136/thx.52.12.1081
- D. P. Strachan and D. G. Cook, “Parental Smoking and Lower Respiratory Illness in Infancy and Early Childhood,” Thorax, Vol. 52, No. 10, 1997, pp. 905-914. doi:10.1136/thx.52.10.905
- International Agency for Research on Cancer (IARC), “Monographs on the Evaluation of Carcinogenic Risk of Chemicals on Humans,” Tobacco Smoking and Tobacco Smoke, Vol. 83, 2002. http://monographs.iarc.fr/htdocs/monographs/vol/83/01-smoking.html
- R. L. Heath and L. Packer, “Photoperoxidation in Isolated Chloroplasts, 1. Kinetics and Stoicheistry of Fatty Acid Peroxidation,” Archieves of Biochemistry and Biophysics, Vol. 125, No. 3, 1968, pp. 189-190. doi:10.1016/0003-9861(68)90654-1
- H. K. Prins and J. A. Loos, “Glutathione,” In: J. G. Yunis, Ed., Biochemical Methods in Red Cell Genetics, Academic Press, New York, 1969, pp. 127-129.
- M. Madesh and K. A. Balasubramanian, “Mirotiter Plate Assay for Superoxide Dismutase Using MIT Reduction by Superoxide,” India Journal of Biochemistry & Biophys, Vol. 35, 1998, pp. 184-188.
- H. E. Aebi, “Catalase,” In: H. U. Bergmeyer, J. Bergmeyer and M. GraBI, Eds., Methods of Enzymatic Analysis, 3rd Edition, Verlag Chemie, Weinheim, 1983, pp. 273- 286.
- D. M. Goldberg and R. J. Spooner, “Glutathione Reductase,” In: H. U. Bergmeyer, J. Bergmeyer and M. GraBI, Eds., Methods of Enzymatic Analysis, 3rd Edition, Verlag Chemie, Weinheim, 1983, pp. 258-265.
- J. Eritsland, “Safety Considerations of Polyunsaturated Fatty Acids,” American Journal of Clinical Nutrition, Vol. 71, No. 1, 2000, pp. 197S-201S.
- G. Van Ginkel and A. Sevanian, “Lipid PeroxidationInduced Membrane Structural Alterations,” Methods Enzymology, Vol. 233, 1994, pp. 273-288. doi:10.1016/S0076-6879(94)33031-X
- J. Cotovo, L. Onno, P. Justine, S. Lamure and P. Catroux. “Generation of Oxidative Stress in Human Cutaneous Models Following in Vitro Ozone Exposure,” Toxicology in Vitro, Vol. 15, No. 4-5, 2001, pp. 357-362. doi:10.1016/S0887-2333(01)00036-4
- Y. G. Zhu, J. F. Zhou, W. Y. Shan, P. S. Zhou and G. Z. Tong, “Potential Oxidative Stress in the Bodies of Electric Arc Welding Operators: Effect of Photochemical Smog,” Biomedical and Environmental Science, Vol. 17, No. 4, 2004, pp. 381-384.
- Y. Kono and I. Fridovich, “Superoxide Radical Inhibits Catalase,” The Journal of Biological Chemistry, Vol. 257, No. 10, 1982, pp. 5751-5754.
- P. M. Sinet and P. Garber, “Inactivation of Human Cu-Zn Superoxide Dismutase during Exposure to O2 and H2O2,” Archives of Biochemistry and Biophysics, Vol. 212, No. 2, 1981, pp. 411-416. doi:10.1016/0003-9861(81)90382-9
- J. Bornholdt, M. Dybdahl, U. Vogel, M. Hansen, S. Loft, and H. Wallin, “Inhalation of Ozone Induces DNA Strand Breaks and Inflammation in Mice,” Mutation Research, Vol. 520, No. 1, 2002, pp. 63-71.
- A. C. Achudume, “Environmental Health, Development and Economic Empowerment Ofrural Women in Nigeria,” Environment, Development and Sustainability, Vol. 11, No. 2, 2009, pp. 459-469. doi:10.1007/s10668-007-9124-1
- Strait and IARC Monograph Working Group, “Carcinogenity of Some Indoor Pollutants: Emissions from Household Combustion of Bioass Fuel, and High-Temperature Frying,” Lancet Oncology, Vol. 7, 2006, pp. 628-629.
- P. Find, G. R. Cass and B. R. T. Simoneit, “Chemical Characterization of Fine Particle Emissions from the Fireplace Combustion of Woods Grown in the Southern United States,” Environmental Science and Technology, Vol. 36, No. 7, 2002, pp. 1442-1451. doi:10.1021/es0108988
- K. A. Miller, D. S. Siscovik, L. Sheppard, K. Sheppard, J. H. Sullivan, G. L. Anderson and J. D. Kaufman, “LongTerm Exposure to Air Pollution and Incidence of Cardiovascular Events in Woman,” The New England Journal of Medicine, Vol. 356, No. 5, 2007, pp. 447-458. doi:10.1056/NEJMoa054409
- K. R. Smith, “Health Impact of Household Fuelwood Use in Developing Countries,” Unasylva, Vol. 224, No. 54, 2006, pp. 41-44.
- J. Zhang and K. R. Smith, “Household Air Pollution from Coal and Biomass Fuels in China: Measurement, Health Impacts and Interventions,” Environmental Health Perspective, Vol. 115, 2007, pp. 848-855.
NOTES
*Corresponding author.