Case Reports in Clinical Medicine
Vol.3 No.4(2014), Article ID:44511,5 pages DOI:10.4236/crcm.2014.34049
γδ-T Large Granular Lymphocyte Leukemia Associated Hemaphagocytic Syndrome Complicated with Multiple Organ Dysfunction
Suigui Wan, Chengcheng Zheng, Yang Lin, Hong Zhao, Li Su, Changqing Xia*
Department of Hematology, Xuanwu Hospital, Capital Medical University, Beijing, China
Email: *cqx65@yahoo.com
Copyright © 2014 by authors and Scientific Research Publishing Inc.
This work is licensed under the Creative Commons Attribution International License (CC BY).
http://creativecommons.org/licenses/by/4.0/



Received 13 January 2014; revised 9 February 2014; accepted 6 March 2014
ABSTRACT
Hemophagocytic syndrome (HPS) usually presents as a complication of T/NK cell lymphoma. However, γδ-T large granular lymphocyte leukemia (LGL) associated HPS was rarely reported. Herein, we reported a case of γδ-T LGL associated HPS. A previously healthy 21-year-old Chinese man was admitted with high fever, severe pancytopenia and liver dysfunction. He developed disseminated intravascular coaggulation (DIC), shock, gastrointestinal bleeding, acute renal failure as well as central nervous system involvement. Bone marrow aspiration showed infiltration of large granular lymphocytes and hemophagocytic histiocytes. Immunophenotyping analysis by four-color flow cytometry showed the leukemic cells with large side scatter (SSC), CD3+, CD2+, CD8+, CD5−, CD7 partly+, CD4−, CD56−, CD57−. Polymerase chain reaction (PCR) amplification of TCRβ and γ chain gene rearrangement confirmed TCRγδ T cell clone. Thus, a diagnosis of γδ-T LGL associated HPS was established. The patient worsened rapidly and died of multiple organ failure.
Keywords:γδ-T Large Granular Lymphocyte Leukemia, Hemophagocytic Syndrome, Multiple Organ Failure

1. Introduction
Hemophagocytic syndrome (HPS) is a rare disorder characterized by the clinical features of fever, pancytopenia, hepatosplenomegaly, and liver dysfunctiona. Which is a clinicopathological entity characterized by increased proliferation and activation of benign macrophages with hemophagocytosis throughout the reticuloendothelial system. HPS can be developed as a rare but potentially fatal complication of several disorders including malignancies, infections, and autoimmune disorders. γδ-T large granular lymphocyte leukemia is particularly rare. Tanaka et al. reported a case of γδ-T large granular lymphocyte leukemia preceded by pure read cell aplasia and complicated with hemophagocytic syndrome causesd by Epstein-Barr Virus infection [1] . Herein, we reported an unusual case of HPS with rapid evolution of multiple organ failure preceded by γδ-T large granular lymphocyte leukemia, and its prevalence, pathogenesis, diagnosis and treatment were discussed.
2. Case Report
A previously healthy 21-year-old Chinese man was admitted to our department with a 6-day history of discontinuous high fever, pancytopenia and liver dysfunction. There was no other complaint and no history of infectious diseases and systemic diseases. Physical examination showed the temperature was 39.7˚C, the blood pressure was 110/70 mmHg, the pulse rate was 110 min–1 and respiratory rate was 30 min–1. He had jaundice, small palpable cervical, groin lymph nodes, and slight hepatomegaly. Upon admission, his total white cell count was 1.58 × 109/L, platelet count was 10 × 109/L. His hemoglobin level started to fall from 140 g/L to 77 g/L. Fibrinogen was 1.53 g/l (NR: 2 - 4 g/L). International normalized ratio (INR) and prothrombin time (PT) were 1.8 (NR: 0.8 - 1.2) and 20.8 (NR: 11 - 15 s), respectively. Activated partial thromboplastin time (APTT) was elevated 123.4 s (INR: 29.1 s). D-Dimer level was 10.9 mg/L (NR: 0 - 0.4 mg/L) C-reactive protein was 14.1 mg/dL (NR: 0.1 - 0.8 mg/dL). His levels of Alanine Aminotransferase (ALT), aspartate aminotransferase (AST), and lactate dehydrogenase (LDH) were elevated to 323 IU/L (NR: 8 - 40 IU/L), 1126 IU/L (NR: 8 - 40 IU/L), and 6099 IU/L (NR: 109 - 245 IU/L), respectively. Total bilirubin (TBIL) and direct bilirubin (DBIL) were 105.3 μmol/L (NR: 3.42 - 23.34 μmol/L) and 92.2 μmol/L (NR: 0 - 8.24 μmol/L), respectively (Table 1). The serum tests for hepatitis A, B, C, E, human immunodeficiency virus, and Anti-treponema pallidum were all negative. Serum Ferritin was 461.4 ng/ml (23.9 - 336.2 ng/ml). The computerized tomography scan for chest and abdomen was performed and indicated a small amount of pleural effusion on both sides, bilateral pulmonary infection, hepatomegaly, a small amount of ascites, abdominal and groin lymph nodes enlargement. On admission, the patient was treated by broad spectrum antibiotics (imipenem/vancomycin) and blood component transfusion. On the second day after admission, bone marrow aspirate was performed and revealed hypoplastic marrow with hemophagocytic histocytes (Figure 1). Immunophenotyping analysis by four-color flow cytometry showed 17%

Table 1. Laboratory findings at different time points.
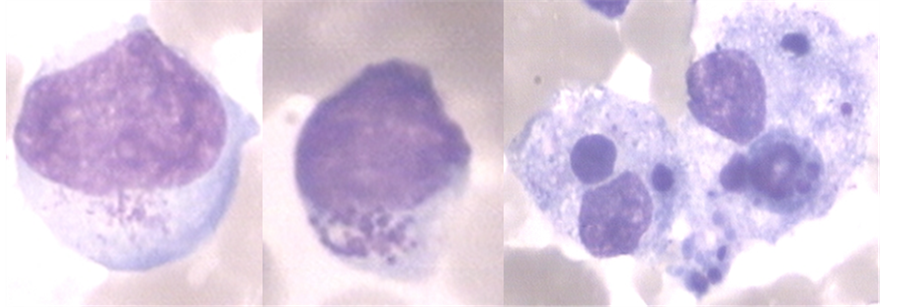
Figure 1. Bone marrow smear showing LGL and macrophage with hemophagocytosis (Wright Stain, ×1000). The left two images display LGL cells with large granular particles in the cytoplasm. The right images exhibit activated macrophages with phagocytosis of blood cells.
lymphocytes in total bone marrow nucleated cells expressed large SSC, CD3+, CD2+, CD8+, CD5−, CD7 partly+, CD4−, CD56−, CD57− (Figure 2). The diagnosis was considered to be T-LGL-associated Hemophagocytic syndrome, disseminated intravascular coagulation (DIC). High dose methylprednisolone was commenced. However, his fever failed to resolve and he quickly developed shock, gastrointestinal bleeding, microscopic hematuria and altered mental status during the next 24 hours. He died of multiple organ failure on the fourth day after admission despite intensive support due. The results of polymerase chain reaction (PCR) amplification of TCR gamma chain gene rearrangement came back and demonstrated a TCR gamma chain rearrangement.
3. Discussion
In this case report, we described a rare case of γδ T cell leukemia proceeding with aggressive HPS. The clinical manifestations of this case are largely attributed to the accompanied HPS. The quick worsening of his clinical condition indicates the aggressiveness of the disease. This is probably due to the activation of the malignant clonal γδ T cells, which can produce high levels of pro-inflammatory cytokines causing dysfunction or even failure of multiple organs as described in this case. γδ T cell leukemia is mostly described in case reports. There have been no available data on its prevalence in certain populations. Its diagnosis is dependent on marrow cell morphology, flow cytometric analysis as well as PCR detection of TCR γ and δ chain gene rearrangement. γδ T cell leukemia is treated by chemotherapy using CHOP protocol. The prognosis depends on the progression and response to chemotherapy of the disease [1] [2] .
Hemophagocytic syndrome (HPS) is a rare but life-threatening disease caused by inappropriate activation of T-lymphocytes and histocytes, hypercytokinemia and hemophagocytosis. The most common symptoms are fever, hepatosplenomegaly, pancytopenia, coagulopathy, hyperferritinemia and lipid abnormalities (elevated triglyceride) [3] . HPS is classified into two forms: primary, inherited (Familial Hamophagocytic Lymphohistocytosis, FHL) and secondary (associated with infection [4] [5] , malignancy [6] , autoimmune disease [7] [8] . HLH Secondary to malignancy occurs most commonly in T or NK-cell lymphoid neoplasms. B-cell malignancies are less common complicated with HLH [9] . Although the diagnostic guidelines have been available [10] , because of its rarity, it is often unrecognized in the clinic. The overall prognosis of HPS is poor, but varies depending on the underlying disease and aggressiveness [11] . Early diagnosis and appropriate treatment is crucial for the management of this disease. However, many patients die because of the aggressive nature of the disease. The case presented in this report died of multiple organ failure because of rapid progressing and uncontrollable HPS.
HPS usually presents as a complication of malignant tumors such as lymphoma. It is common that large granular NK cell leukemia leads to HPS because leukemic NK cells secrete high levels of pro-inflammatory cytokines subsequently activating macrophages which in turn phagocytose blood cells causing HPS. NK cell granular leukemic cells express CD57 but are negative for CD3 and TCR. γδ-T large granular lymphocyte leukemia resembles NK granular lymphocyte leukemia morphologically, but the leukemic cells express T cell markers and γδ TCRs. These T cells can produce large amount of cytokines after activation [12] . γδ-T large granular lymphocytes leukemia is usually diagnosed by flow cytometry showing clonal γδ T cells, or through polymerase
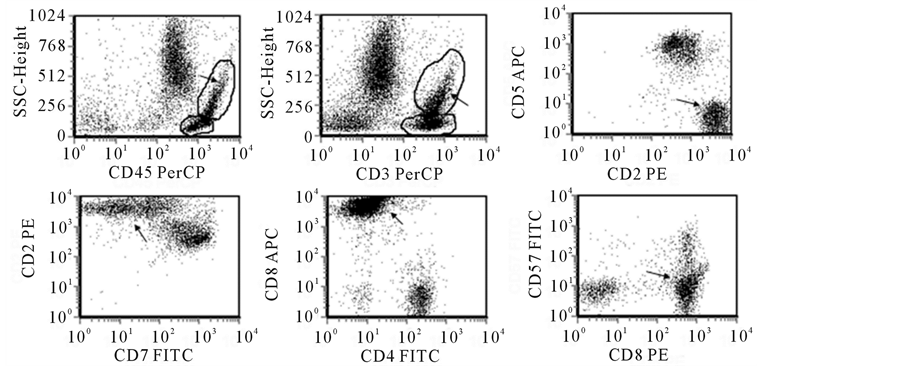
Figure 2. Immunophenotype of bone marrow cells examined using 4-color flow cytometry. Bone marrow cells were stained with a panel of fluorescent antibodies against a variety of T cell markers indicated in the above flow plots, and analyzed using FCS express software using different combinations. The population pointed by the arrow in each plot is the LGL cell population.
chain reaction (PCR) for γ chain gene rearrangement. This patient is a γδ T granular lymphocyte leukemia diagnosed by PCR for γ chain gene rearrangement. The case was positive for CD8 but negative for CD4. γδ T granular lymphocyte leukemia usually shows aggressive nature clinically with unclear mechanism. The prognosis of γδ T granular lymphocyte leukemia complicated with HPS is extremely poor. It has been documented that serum fibrinogen level is an independent adverse prognostic factor. Early diagnosis is, therefore mandatory, the intensive care should be commenced early, and new treatments including bone marrow transplantation should be considered [12] .
Because of the hyperactivation state of the global immune system, treatment should be surrounding immune suppression. Treatment options include chemotherapy usually using CHOP protocol, plasma exchange, immunosuppressants such as cyclosporine, as well as hematopoietic stem cell transplantation. Because of the elevated levels of pro-inflammatory cytokines such as TNF-α and IFN-γ, which may largely contribute to the progression of the disease, plasma exchange should be considered early to remove those detrimental factors. Overall, γδ T granular lymphocyte leukemia is a rare condition especially complicated with HPS. The prognosis is poor, which may be improved by early diagnosis and intensive effective management.
References
- Tanaka, Y., Matsui, K., Yamashita, K., et al. (2006) T-γδ Large Granular Lymphocyte Leukemia Preceded by Pure Red Cell Aplasia and Complicated with Hemophagocytic Syndrome Caused by Epstein-Barr Virus Infection. Internal Medicine, 45, 631-635. http://dx.doi.org/10.2169/internalmedicine.45.1594
- Bourgault-Rouxel, A.S., Loughran Jr., T.P., Zambello, R., et al. (2008) Clinical Spectrum of Gammadelta+ T Cell LGL Leukemia: Analysis of 20 Cases. Leukemia Research, 32, 45-48. http://dx.doi.org/10.1016/j.leukres.2007.04.011
- Janka, G.E. (2007) Hemophagocytic Syndromes. Blood Reviews, 21, 245-253. http://dx.doi.org/10.1016/j.blre.2007.05.001
- Phillips, J., Staszewski, H. and Garrison, M. (2008) Successful Treatment of Secondary Hemophagocytic Lymphohistiocytosis in a Patient with Disseminated Histoplasmosis. Hematology, 13, 282-285. http://dx.doi.org/10.1179/102453308X316013
- Imashuku, S., Kuriyama, K., Sakai, R., et al. (2003) Treatment of Epstein-Barr Virus-Associated Hemophagocytic Lymphohistiocytosis (EBV-HLH) in Young Adults: A Report from the HLH Study Center. Medical and Pediatric Oncology, 41, 103-109. http://dx.doi.org/10.1002/mpo.10314
- Celkan, T., Berrak, S., Kazanci, E., et al. (2009) Malignancy-Associated Hemophagocytic Lymphohistiocytosis in Pediatric Cases: A Multicenter Study from Turkey. The Turkish Journal of Pediatrics, 51, 207-213.
- Cortis, E. and Insalaco, A. (2006) Macrophage Activation Syndrome in Juvenile Idiopathic Arthritis. Acta Paediatrica Supplement, 95, 38-41. http://dx.doi.org/10.1080/08035320600649713
- Parodi, A., Davi, S., Pringe, A.B., et al. (2009) Macrophage Activation Syndrome in Juvenile Systemic Lupus Erythematosus: A Multinational Multicenter Study of Thirty-Eight Patients. Arthritis & Rheumatology, 60, 3388-3399. http://dx.doi.org/10.1002/art.24883
- Han, A.R., Lee, H.R., Park, B.B., et al. (2007) Lymphoma-Associated Hemophagocytic Syndrome: Clinical Features and Treatment Outcome. Annals of Hematology, 86, 493-498. http://dx.doi.org/10.1007/s00277-007-0278-6
- Henter, J.I., Horne, A., Aricò, M., et al. (2007) HLH-2004: Diagnostic and Therapeutic Guidelines for Hemophagocytic Lymphohistiocytosis. Pediatric Blood & Cancer, 48, 124-131. http://dx.doi.org/10.1002/pbc.21039
- Trottestam, H., Horne, A., Aricò, M., et al. (2011) Chemoimmunotherapy for Hemophagocytic Lymphohistiocytosis: Long-Term Results of the HLH-94 Treatment Protocol. Blood, 118, 4577-4584. http://dx.doi.org/10.1182/blood-2011-06-356261
- Shabbir, M., Lucas, J., Lazarchick, J., et al. (2011) Secondary Hemophagocytic Syndrome in Adults: A Case Series of 18 Patients in a Single Institution and a Review of Literature. Hematological Oncology, 29, 100-106. http://dx.doi.org/10.1002/hon.960
Abbreviations
HPS: Hemophagocytic syndrome LGL: Large granular lymphocyte leukemia DIC: Disseminated intravascular coaggulation SSC: Side scatter PCR: Polymerase chain reaction INR: International normalized ratio PT: Prothrombin time APTT: Activated partial thromboplastin time ALT: Alanine aminotransferase AST: Aspartate aminotransferase LDH: Lactate dehydrogenase TBIL: Total bilirubin DBIL: Direct bilirubin FHL: Familial hamophagocytic lymphohistocytosis
NOTES

*Corresponding author.