Modern Research in Catalysis
Vol.2 No.2(2013), Article ID:30545,9 pages DOI:10.4236/mrc.2013.22008
Cationic Metalloporphyrins Imobilized in Faujasite Zeolites as a Cytochrome P-450 Mimic
1Departamento de Química, FFCLRP, Universidade de São Paulo, Ribeirão Preto, Brazil
2Departamento de Química, Universidade Federal de São Carlos, São Carlos, Brazil
3Instituto de Química, Universidade de São Paulo, São Paulo, Brazil
Email: *ilvrosa@ufscar.br
Copyright © 2013 Luciana P. B. Lôvo et al. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Received December 1, 2012; revised January 10, 2013; accepted January 21, 2013
Keywords: Metalloporphyrin; Iodosylbenzene; Hydrocarbon Oxidation; Y Zeolite
ABSTRACT
Metalloporphyrins immobilized into NaY zeolite are described as catalysts for hydrocarbon oxyfuntionalization. Manganese(III) and iron(III)tetrakis(4-N-methylpyridyl)-porphyrin (MnP1 and FeP1), and manganese(III) and iron(III) tetrakis(4-N-benzylpiridil)-porphyrin (MnP2 and FeP2) were impregnated (MnP1-NaYimp, FeP1-NaYimp, MnP2-NaYimp, FeP2-NaYimp, respectively) and encapsulated (MnP1-NaY, FeP1-NaY, MnP2-NaY and FeP2-NaY) into the NaY zeolite. These catalysts were used in the oxidation of (Z)-cyclooctene, cyclohexane, and adamantane by iodosylbezene (PhIO). These systems were able to epoxidize (Z)-cyclooctene with cis-epoxycyclooctane yields as high as 100%. By using cyclohexane and adamantane as substrate, the susceptibility of the benzyl groups on the porphyrin ring of the MnP materials, led to a different distribution of the oxidized products. With FePs, this susceptibility was not detected because the species responsible for the oxidations, FeIV(O)P•+, is more active than MnV(O)P. In conclusion, cationic metalloporphyrins immobilized into NaX zeolites, are good cytochrome P-450 models is less polar solvents since the selectivity of the system indicates the “in cage” solvent oxygen rebound oxidative process.
1. Introduction
The superfamily of cytochrome P-450 enzymes plays an important role in oxidative processes in nature. Due to their importance, a large number of publications have been produced since the early 1950-60’s [1-4]. The evidences of the oxidation processes, chemistry, structure, and mechanism of cytochrome P450 continue to be outstanding in recent reviews [5-10].
A large number of studies using synthetic analogues of cytochrome P-450 as biomimetic or bioinspired catalysts for the selective oxidation of organic molecules have been developed [11-23]. The design of efficient catalysts that are stable in the reaction media and promote selective hydrocarbon oxidation under mild conditions with high turnovers has been a challenge. However, a problem associated with hydrocarbon oxidation catalyzed by metalloporphyrins (MePs) is the oxidative self-destruction of the catalyst due to the oxidizing media [2,8]. In this sense, it is difficult to perform the separation of the metalloporphyrin (MeP) from the reaction mixture and to reuse it, since it is soluble in polar media.
One approach to solve this problem is the use of electron withdrawing substituents on the porphyrin periphery, especially halogenated and perhalogenated phenyl porphyrins, thus producing more robust and resistant catalysts [5,6,9-11]. Another solution to promoting the stability of MeP catalysts is their immobilization on a solid matrix. In general, immobilization methods include physical entrapment or entrapment through covalent binding. Catalyst immobilization onto solid surfaces has been achieved through electrostatic interactions and covalent binding [24-32]. In the case of cationic MePs immobilization can occur simply through electrostatic interactions between these catalysts and an inorganic matrix such as zeolite [14-18].
The steric and electronic effects in synthetic models are responsible for the large number of studies on their stability and selectivity in hydrocarbon biomimetical oxidation [33-36]. In this context, the use of zeolites in an attempt to mimic the protein cavity of natural enzymes is a good alternative, since its nanopores and nanochannels are a three-dimensional network of well-defined crystalline structures, thus conferring the shape and size selectivity [37]. These well-organized nanopores and nanochannels of the zeolites provide a controlled steric environment for the MeP, acting as a model for the active site of cytochrome P450 [14]. Moreover, MeP immobilization enables recovery and re-use [38,39].
The impregnation and encapsulation of these catalysts into the pores of mesoporous molecular sieves have introduced new opportunities for the design of new porphyrin/carrier systems. Site isolation of the metal active species is one of the most attractive methods, and it consists in the hydrothermal synthesis of faujasite-type zeolites in the presence of cationic MePs, the so-called shipin-a-bottle system [40]. In this approach, the cationic MeP is stabilized inside the zeolite nanocages and is physically trapped into the inorganic matrix.
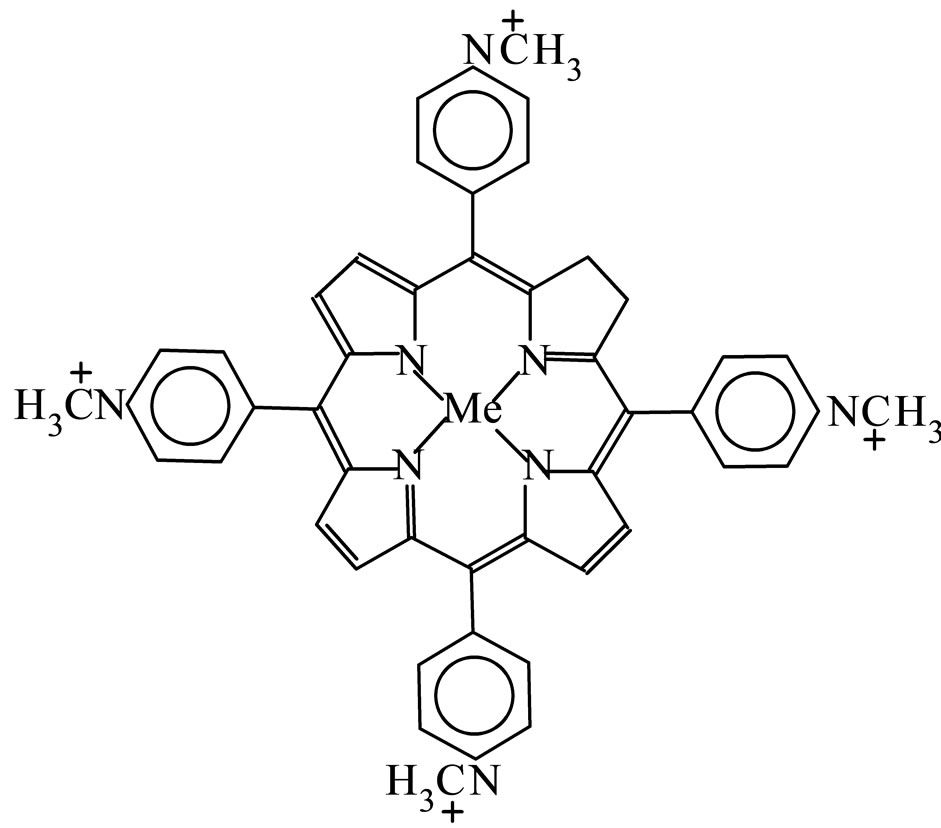
In this work, the iron and manganese(III)tetrakis(4-Nmethylpiridil)porphyrin (FeP1 and MnP1) as well as iron and manganese(III)tetrakis(4-N-benzylpiridil) porphyrin (FeP2 and MnP2), presented in Figure 1, were encapsulated in the zeolite NaY, to leading the heterogeneous catalysts FeP1-NaY, MnP1-NaY, FeP2-NaY, and MnP2- NaY, respectively. Moreover, the MePs were also impregnated in the zeolite surface to obtain FeP1-NaYimp, MnP1-NaYimp, FeP2-NaYimp and MnP2-NaYimp, respectively, for comparison purposes.
All these materials were characterized by UV-Vis spectroscopy, powder X-ray diffraction (XRD), thermogravimetric analysis (TGA/DTA), scanning electronmicroscopy (SEM), energy dispersive X-ray spectrometry (EDS), and Al27 MAS NMR. Some of these characterizations have been previously described for FeP1-NaY, FeP2-NaY and MnP1-NaY [14,41,42]. The oxidation of (Z)-cyclooctene, cyclohexane, and adamantane catalyzed by these materials were studied using iodosylbenzene (PhIO) as the oxidizing agent in solvent media with different polarities was accomplished in order to evaluate the reaction mechanism in each of the studied systems.
2. Experimental
2.1. FeP-NaYimp and MnP-NaYimp Synthesis
MeP impregnation into the solid support was achieved by stirring a MeP solution in H2O with a suspension of NaY for 50 min [43]. The resulting catalysts FeP-NaYimp and MnP-NaYimp were washed with water, methanol, and 1,2 - dich loroethane in a Soxhlet extractor overnight, to remove unbound and weakly bound MeP. The solids were dried at 60˚C for 24 h.
2.2. FeP-NaY and MnP-NaY Synthesis
NaY zeolites were synthesized in the presence of iron and
 (a)
(a)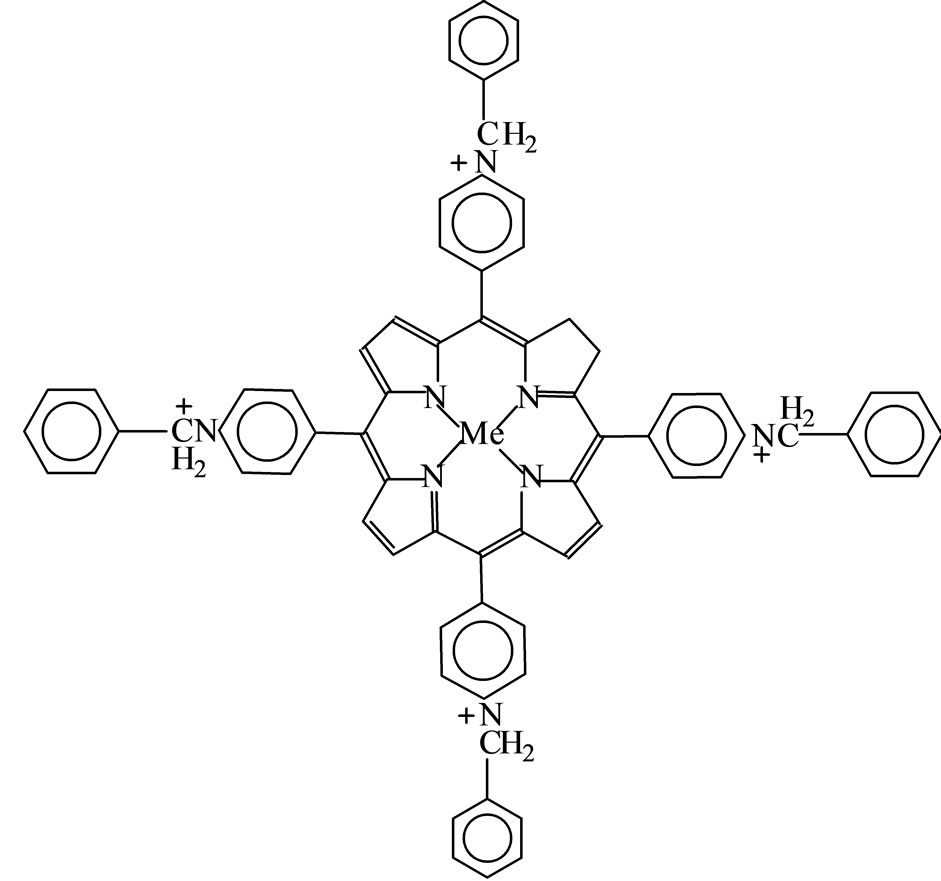 (b)
(b)
(a): Iron and manganese(III)tetrakis
(4-N-methylpyridyl)porphyrin (FeP1 and MnP1)
(b): Iron and manganese(III)tetrakis
(4-N-benzylpyridyl) porphyrin (FeP2 and MnP2)
Figure 1. Metalloporphyrins represented by 1) MnP solu- tion or FeP solution when as a pure solid or in solution; 2) MnP-NaYimp or FeP-NaYimp when impregnated; 3) MnP- NaY or FeP-NaY when encapsulated.
manganese(III)tetra(4-N-methylpyridyl)porphyrin (FeP1 and MnP1) as well as iron and manganese(III) tetra (4- N-benzylpyridyl)porphyrin (FeP2 and MnP2), to obtain the encapsulated catalysts FeP1-NaY, MnP1-NaY, FeP2- NaY, and MnP2-NaY, respectively. According to an already reported procedure [40-42], the inclusion of cationic MePs inside the nanocages of faujasite NaY can be performed by hydrothermal synthesis. In the present case, a silicate gel was prepared by stirring 0.70 g of silica, 0.60 g of NaOH, 3.6 × 10−5 mol of the desired MeP and 1.5 ml H2O. The gel was then added to an aluminate solution consisting in 1.55 g of aluminum isopropoxide (Al[(CH3)2-CHO]3), 0.65 g of NaOH, and 2.0 ml of H2O. This mixture was transferred to a polypropylene bottle containing 6.0 ml of H2O and stirred at room temperature for 24 h. The bottle was placed in a water bath at 90˚C for 15 h, and then 20 ml of H2O was added. The resulting solids were filtered, washed several times with water, and dried at 80˚C for 24 h. The samples were Soxhletextracted with water for 3 days, and then with methanol for another 3 days, to remove all the MeP present on the external surface of the zeolite. The samples were dried at 80˚C for 24 h.
2.3. Catalysts Characterization
Powder X-ray diffraction (XRD) of the samples was carried out on a Siemens D 5005 diffractometer using CuKa X-radiation. Variable temperature in situ powder XRD was carried out using an Anton Paar HTK16 sample chamber in a Bruker-AXS D8 Advance diffracttometer with a Braun position sensitive detector. The samples were heated at 10˚/min until 1000˚C under air and kept at this temperature for 10 min. The samples were treated with hot concentrated hydrochloric acid, to destroy the inorganic framework before recording the UV-Vis spectra. No demetallation was observed for the organic macrocycle. The thermogravimetric (TGA/DTA) curves were obtained using a TA Instruments STD 2690. Scanning electron microscopy (SEM) and energy dispersive X-ray spectrometry (EDS) were carried out on a Zeiss EVO 50 microscope. 27Al solid-state magic angle spinning NMR spectra were recorded on a Varian model UNIT-Plus, 9.4 Tesla spectrometer.
2.4. Catalytic Reactions
The reactions were carried out in a 4 ml vial sealed with a teflon-coated silicone septum. A total of 2.02 × 10−2 mol of solvent (1,2-dichloroethane or toluene), 1.46 × 10−2 mol of cyclohexane or 8.22 × 10−1 mol of (Z)- cyclooctene or 8.90 × 10−4 mol of adamantine, and the internal standard (bromobenzene in the case of cyclohexane and (Z)-cyclooctene oxidations; cyclopeantanone for adamantane oxidation) were added to the vial containing 2.5 × 10−7 mol of MeP and 5.0 × 10−6 mol of the oxidant agent, PhIO. The mixture was stirred at room temperature for 24 hours. The products were analyzed by gas chromatography using a Hewlett Packard GC System HP 6890 Series equipped with a FID detector and a megabore column (30 m × 0.25 mm); nitrogen was employed as the carrier gas. The product yields were based on the initial concentration of PhIO.
2.5. Catalyst Reuse and Stability
The stability of the catalyst was studied by performing repeated oxidation reactions using the same reaction conditions as described above. (Z)-cyclooctene was used as model substrate. At the end of each reaction cycle, the catalyst was recovered by filtration and washed with water and DCE, dried, and reused. The conversion (%) was almost identical, irrespective of the number of cycles. No evidence of catalyst decomposition was observed during the catalytic reaction and no MeP could be detected by spectroscopic UV-Vis measurement of the liquid reaction mixture after each catalytic reaction.
3. Results and Discussion
3.1. Catalysts Characterization
The catalysts presented good X-ray powder diffraction patterns, showing narrow peaks characteristic of the main planes of the NaY zeolite [44], where 2q = 27.1˚; 30.0˚, and 34.2˚ for the (5 3 3), (6 4 2) and (7 5 1) planes, respectively (in Figures 2(A)-(E)).
The dependence of the lattice parameter on the temperature is shown in Figure 3. The powder patterns were indexed after the JCPDS file 85 - 2064. The influence of the temperature on the inorganic lattice is similar for FeP1-NaY and NaY. The fact that the lattice constant of FeP1-NaY is higher than that of NaY suggests that the framework of the former is more expanded due to the nanoinclusion of the FeP complex [14].
The UV-Vis spectrum of MnP1-NaY is similar to that of MnP1 in aqueous solution. The MnP1 complexes present absorption bands at 366, 456 and 558 nm. Concern-
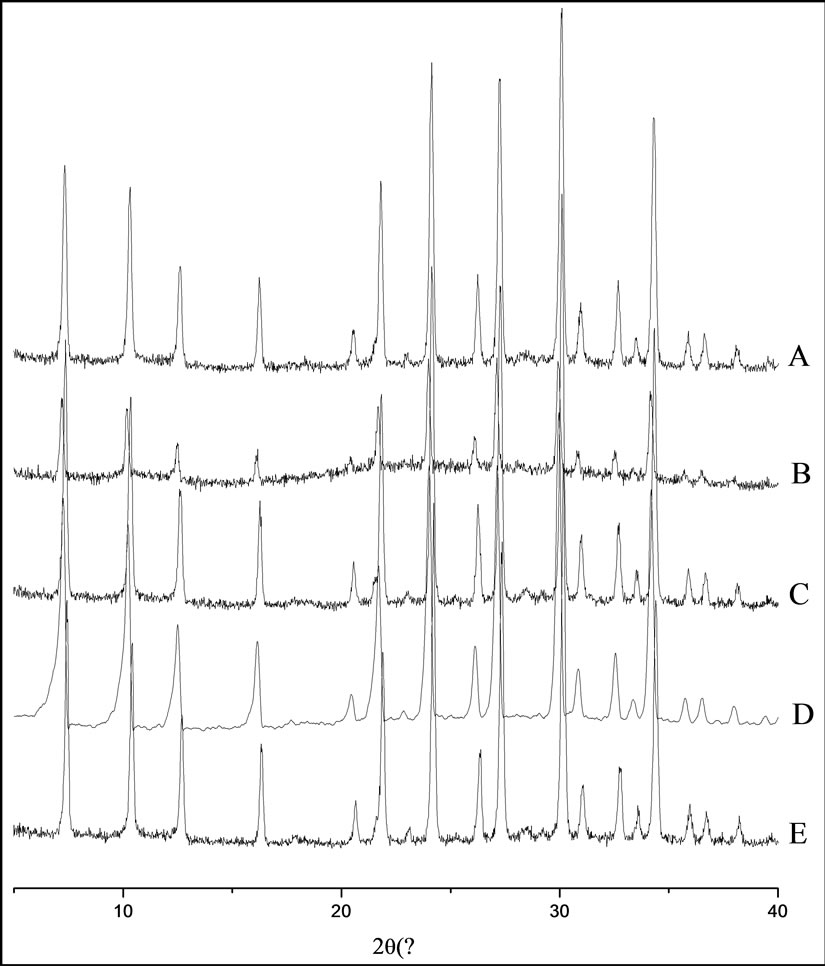
Figure 2. Powder XRD patterns of (A) NaY, (B) Mn[T(4-NMePy)P]-NaY, (C) Mn [T(4-N-MePy)P]-NaYimp, (D) Fe[T- (4-N-MePy)P]-NaY, and (E) Fe[T(4-N-MePy)P]-NaYimp. Temperature 25˚C.
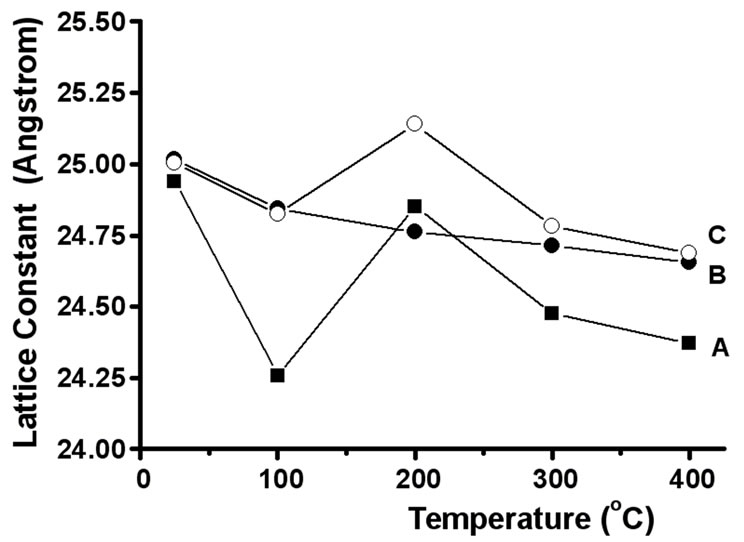
Figure 3. Influence of the temperature on the lattice constant of NaY (A), FeP1-NaYimp (B), and FeP1-NaY (C).
ing the zeolite-encapsulated MnP1, the UV-Vis bands were red-shifted to 396, 472, and 561 nm if compared to the initial MnP1 complexes. This behavior can possibly be ascribed to the distortion of the porphyrin ring after the encapsulation process [45]. The absorbance of the Soret band was used to evaluate the loading of MnP1 per gram of MnP1-NaY, which was 8.15 × 10−8 mol∙g−1.
Through the TGA/DTA analysis (not shown), it was possible to verify that the standard of water loss was affected by the presence of the MnPs in the zeolite cavities. In the sample containing encapsulated MnP, the onset of water loss occurs at around 180˚C and finishes at ~450˚C, while in the sample without the MnP water loss occurred from 180˚C to 250˚C. Considering the low loading values, in the range of 10−7 mols per gram of catalyst, the percentage of MeP in the NaY zeolite is very small. Therefore, TGA/DTA analysis was not efficient to determine the amount of metalloporphyrin immobilized into the NaY zeolite.
The MnP1-NaY SEM image presented in Figure 4 is also similar to that of NaY zeolite, indicating that they have the same morphology. The Si/Al ratio for the samples was ascertained by EDS and evaluated as 1.7. This suggests that the framework around the guest molecule MnP1 is faujasite-Y [18].
27Al MAS NMR analysis gave information about the coordination sphere of this atom. For the NaY sample, a signal in 60 ppm was observed, as shown in Figure 5. This signal is described as being characteristic of a tetrahedral molecular structure [46], in this case AlO4. The same happens for the sample where the MeP is impregnated on the NaY surface only. As for the MeP encapsulated samples, another signal next to 5.3 ppm is observed (Figure 5), which is characteristic of molecular structures where tetrahedral and octahedral aluminum atoms exist.
3.2. (Z)-Cyclooctene Oxidation
(Z)-cyclooctene was chosen for the initial catalytic tests because it is considered a diagnostic substrate that en-


Figure 4. SEM images of NaY (A) and Mn[T(4-N-MePy)P]- NaY (B).
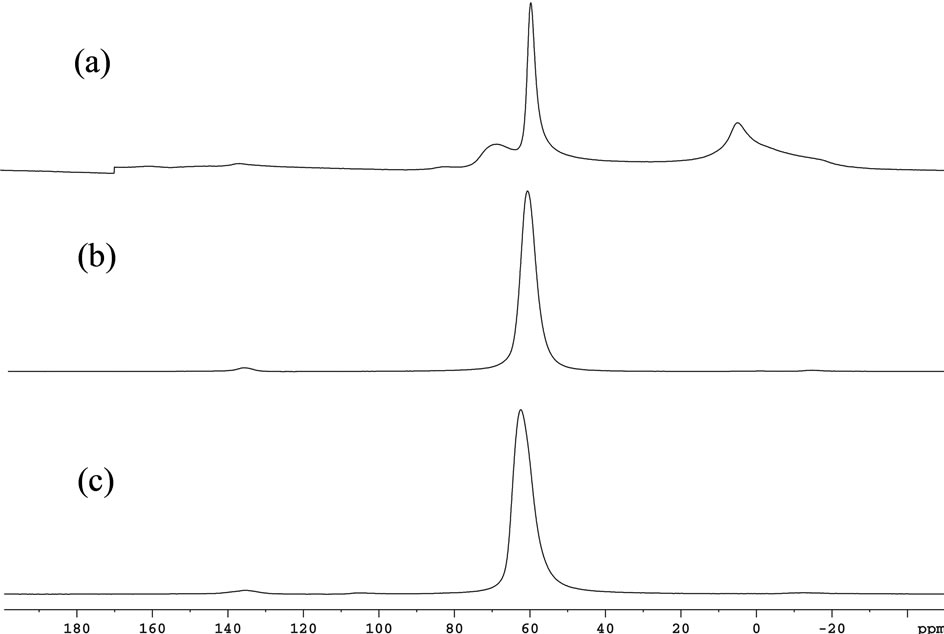
Figure 5. 27Al MAS NMR spectra of (a) MnP1-NaY; (b) MnP1-NaYimp, and (c) NaY zeolite.
ables evaluation of the catalytic potential of a system. Moreover, (Z)-cyclooctene epoxidation by PhIO allows comparison between the efficiency of these impregnated and encapsulated catalysts, and it permits comparison with the analogous homogeneous systems. The results are listed in Table 1.
In homogeneous medium, the catalysts lead to low yields if one considers the very reactive nature of (Z)- cyclooctene, especially in the case of MnP1 (48%, entry 7). This can be attributed to the nature of the MnP. Heterogeneous catalysts are advantageous in all cases, especially the encapsulated systems, since product yields as high as 100% are achieved (Entries 3, 6, 9, 12). These results indicate that the formation of FeIV(O)P•+ or MnV(O)P active species are favored in the zeolite cavities. The control reaction with NaY zeolite alone was carried out, and no product formation was observed.
The encapsulation of MePs makes the catalyst more resistant to oxidative self-destruction [42]. Moreover, it is possible to use these cationic MePs as catalysts for the oxidation of hydrophobic organic substrates in organic medium when they are encapsulated in the zeolite cavities.
3.3. Cyclohexane Oxidation
The main mechanism of alkane hydroxilation reactions is the so-called oxygen rebound which involves hydrogen atom abstraction from the substrate (RH) by a reactive intermediate species, FeIV(O)P•+ or MnV(O)P. These species form a solvent cage, followed by the rapid transfer of the hydroxyl group from FeIV(OH)P or MnIV(OH)P to the cyclohexyl radical (R•), producing the corresponding alcohol [47,48], as represented in Figure 6 [49]. Depending on the MeP system and the polarity of the solvent used in the oxidation reaction, the cage can collapse

Figure 6. Mechanism of oxygen rebound for alkane hydroxylations.
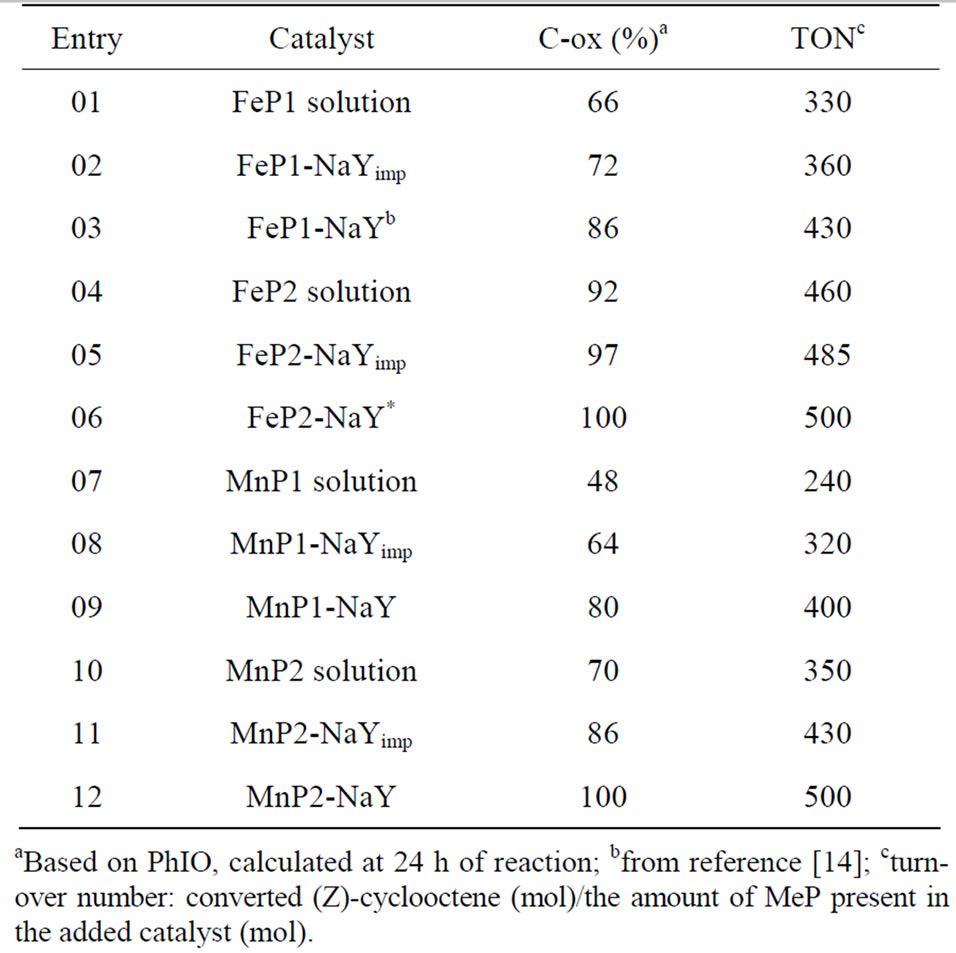
Table 1. Catalytic activity of MnP and FeP systems in the oxidation of (Z)-cyclooctene by PhIO.
and the carbon radicals can escape, leading to the formation of different products, including ketones, as shown in Figure 6 [50].
The catalytic activity of MnP and FeP systems in the oxidation of cyclohexane by PhIO is presented in Table 2. The control reaction with NaY zeolite alone was carried out, and no product formation was detected.
In general, encapsulation increases the total product yield ontained with MeP systems (Entries 15, 19 and 26) when DCE is used as solvent. The total yield is not so high in DCE only in the case of MnP1-NaY (Entry 22). When toluene was used as solvent, cyclohexane was selectively oxidized to cyclohexanol. This is because toluene is less polar (m = 0.37D) than DCE (m = 1.8D), so the “in cage” solvent oxygen rebound oxidative process predominates, giving alcohol as the sole product. As for the MnP2 systems, these catalysts are more selective due to the presence of the benzylic groups (Entries 24-26).
For the FeP1 system, a cyclohexanol yield (C6-ol) of 11% and a cyclohexanone yield (C6-one) of 10% were achieved (Entry 13). So the C6-ol/C6-one ratio was 1.1. When FeP1 was impregnated on the NaY zeolite surface, there was a slight increase in the selectivity of cyclohexane oxidation (Entries 13 and 14). FeP1 encapsulation into the NaY zeolite promoted an increase in the oxidation reaction yield and selectivity (C6-ol yield = 21% and C6-one yield = 7%, Entry 15) for the reaction carried out in DCE. FeP1-NaY was more selective toward the alcohol in toluene, as observed for MnP1-NaY.
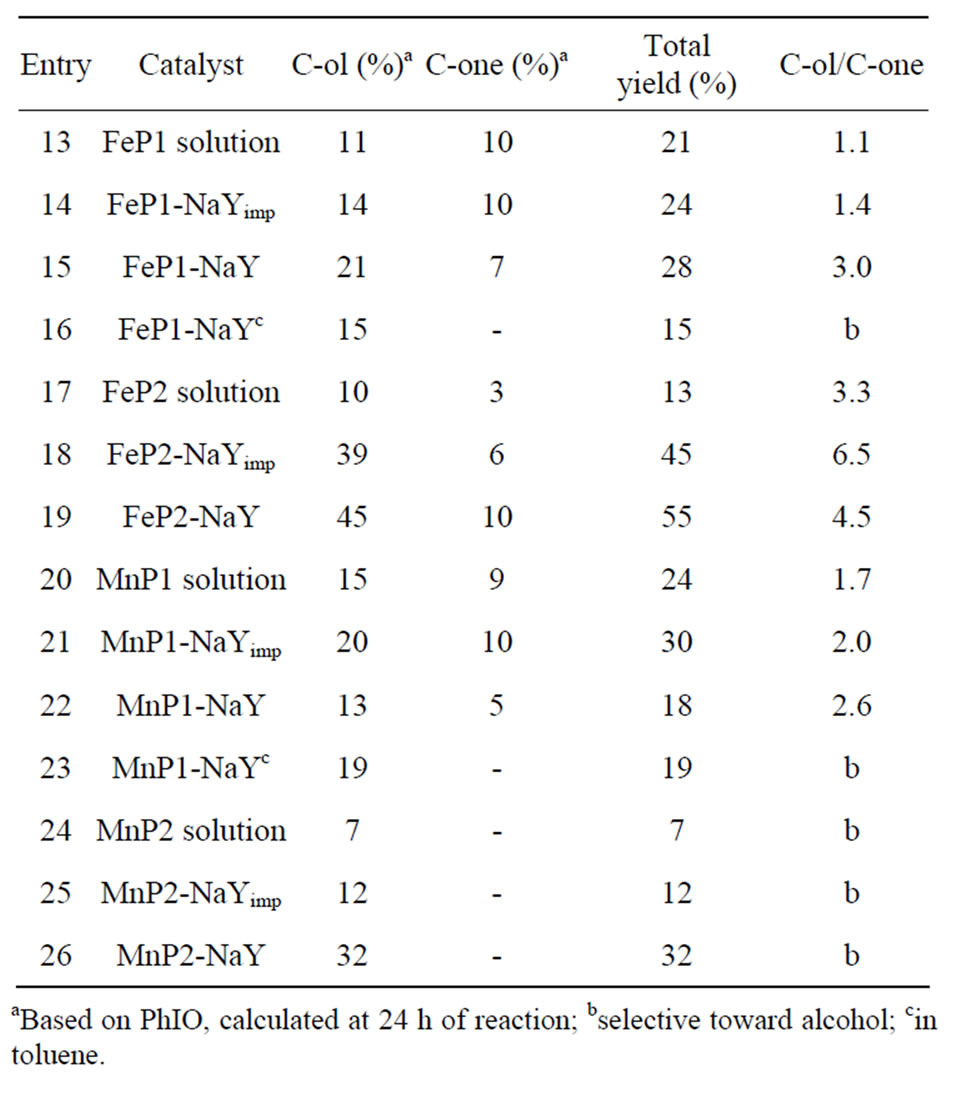
Table 2. Catalytic activity of MnP and FeP systems in the oxidation of cyclohexane by PhIO.
In the case of FeP2 in homogeneous system, the C6-ol yield was 10% and the C6-one yield was 3%, which gives a C6-ol/C6-one ratio of 3.3 (entry 17). FeP2 impregnated on the NaY surface displayed increased selectivity (Entry 18) compared with the homogeneous FeP2 system (Entry 17). When FeP2 was encapsulated into the NaY zeolite, the catalytic system became more selective for the alcohol (C6-ol yield = 45% and C6-one yield = 10%, Entry 19) compared with FeP2 in solution. The benzyl groups present in FeP2 were responsible for the slight small increase in yield and selectivity toward the alcohol, affecting the alkane oxidation mechanism when compared with FeP1.
The encapsulation of FeP, especially FeP2, resulted in a more robust catalyst with higher product yields. Besides, the effect of the zeolite cage favors the oxygen rebound process, so the catalyst acts as a P-450 model, as observed previously for these systems in other solvents [14]. The amount of cyclohexanone produced in the reactions indicates an escape of the intermediate species from the cage, since the carbon radicals can escape from the solvent cage (Figure 6) and react to give a number of other products [50].
The MnP1 systems, which contain methyl pyridyl groups, lead to relatively lower yields. In solution, MnP1 leads to a C6-ol yield of 15% and a C6-one yield of 9% (Entry 20). When MnP1 is impregnated in the zeolite, there is an increase in the total product yield, which is 30% (Entry 21). The encapsulated system gives a C6-ol yield of 13% and a C6-one yield of 5% (Entry 22), showing that the encapsulated MnP1 is a little more selective catalyst than its homogeneous analogue; however, the total yield is lower.
In the case of MnP2 in solution, although the yield is not so high, the oxidation is selective for the alcohol (C6-ol yield = 7%, Entry 24). The heterogeneous catalysts MnP2-NaYimp and MnP2-NaY are more efficient: the C6-ol yields are 12% (Entry 25) and 32% (Entry 26). The effect of MnP2 encapsulation is note worthy, since the C6-ol yield is five times larger than the one obtained with the homogeneous system. Comparison between the MnP1 and MnP2 systems (Entries 20-26) demonstrates that benzyl pyridyl, a flexible and bulky group, gives rise to a more hydrophobic medium, thus favoring alcohol formation and indicating that the oxygen rebound process is operating.
3.4. Adamantane Oxidation
Adamantane is a good diagnostic substrate for mechanistic studies [51,52] since adamantane oxidation can occur at two positions: at the tertiary carbon producing 1- ada-mantanol, and at the secondary carbon, leading to 2-adamantanol and 2-adamantanone (Figure 7). It is well known that adamantane tertiary C-H bonds are more reactive than secondary C-H bonds because they have lower bond energy [51].
The statistically corrected data for adamantane oxidation are presented in Table 3. The control reaction with NaY zeolite alone was carried out, and no products were observed.
FeP2 is a more efficient system for adamantane hydroxylation, since it leads to higher total product yield (Entries 31-33); however, the selectivity is lower. The expected hydroxylation at the tertiary carbon takes place, but there is a significant increase in secondary carbon hydroxylation, accounting for the high total yield. As for the FeP1 system in DCE (Entries 27 - 29), it is more selective toward Ad-1-ol, a tertiary alcohol. When toluene is used as solvent, the oxidation is even more selective, producing exclusively Ad-1-ol (Entry 30). This behavior was also observed in the case of cyclohexane oxidation and it is attributed to the lower polarity of toluene when compared with DCE. The low solvent polarity in this case favors the biomimetic process via the oxygen rebound mechanism.
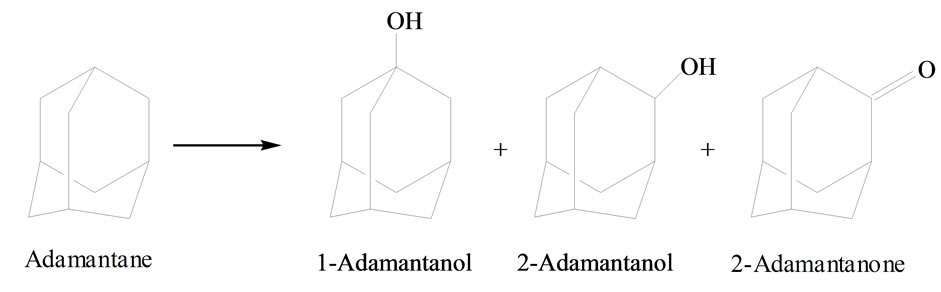
Figure 7. Adamantane and possible oxidized products.
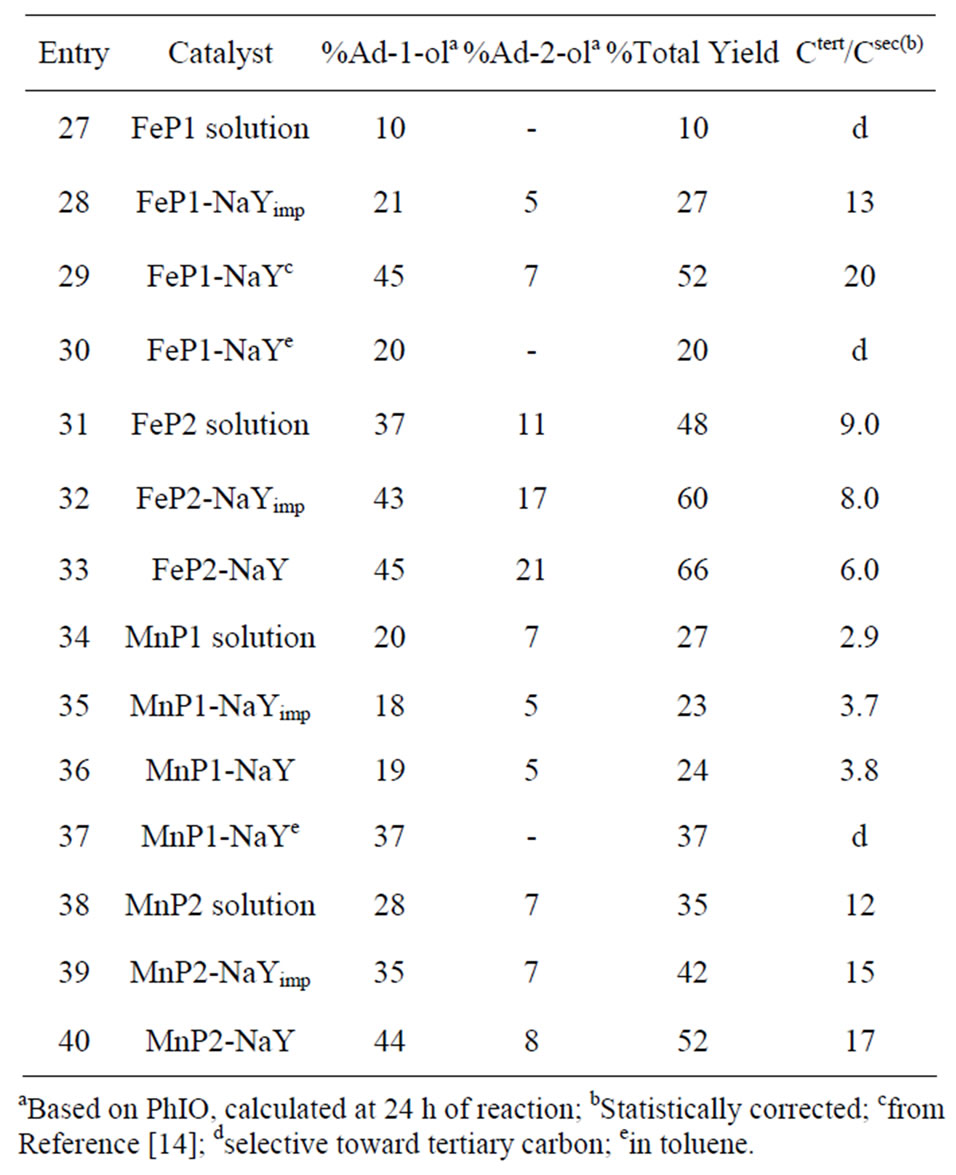
Table 3. Catalytic activity of MnP-NaY and FeP-NaY in the oxidation of adamantane by PhIO.
Encapsulation of MnP1 (Entries 34 - 36) does not favor the oxidation processes when compared with MnP2 (Entries 38-40). MnP1 in DCE (Entry 34), MnP1 impregnated on the zeolite surface (Entry 35), and MnP1 encapsulated in NaY zeolite (Entry 36) lead to the same profile of adamantine oxidation. A total yield between 23% and 27% is observed, and selectivities Ad-1-ol/Ad-2-ol between 2.9 to 3.8 are obtained. However, MnP1-NaY in toluene leads to high Ad-1-ol yield (37% Entry 37) and is selective for this alcohol, indicating that an oxygen rebound process occurred.
Comparing MnP1 (Entry 34) with MnP2 (Entry 38) in DCE solution, the oxidation profile is about the same. This behavior changes significantly when MnP2 is encapsulated into the NaY zeolite. In the latter system, there is an increase in the total yield and selectivity (Entry 40) due to the benzyl groups in MnP2, which are responsible for an increase in total yield and selectivity for Ad-1-ol, as observed in the case of cyclohexane oxidation.
4. Conclusion
Cationic metalloporphyrins immobilized on NaY zeolites are good cytochrome P-450 models since these polar MePs can catalyze hydrocarbon oxidation in the zeolite cavities, in apolar solvent medium, thus mimicking the hydrophobic P450 active sites. The studied encapsulated systems were able to epoxidize (Z)-cyclooctene with cisepoxycyclooctane yields as high as 100%. The use of cyclohexane and adamantane as substrate, demonstrated that the FeIV(O)P•+ species are more active for oxidation than MnV(O)P. This is because the substituent groups (in this case the benzyl groups) do not affect the distribution of oxidized products when the FeP systems are used. However, the substituent groups are more important in the case of MnP systems, since they modify the total yield and selectivities. The behavior of the MeP systems in cyclohexane and adamantane oxidation was similar. However the difference between the observed total yields showed that cyclohexane is a more inert substrate than adamantane.
5. Acknowledgements
This work was supported by CAPES, CNPq and FAPESP.
REFERENCES
- K. Paigen, “Hemoglobin as the Red Pigment of Microsomes,” Biochimica et Biophysica Acta, Vol. 19, No. 2, 1956, pp. 297-299. doi:10.1016/0006-3002(56)90431-0
- M. Klingenberg, “Pigments of Rat Liver Microsomes,” Archives of Biochemistry and Biophysics, Vol. 75, No. 2, 1958, pp. 376-386. doi:10.1016/0003-9861(58)90436-3
- T. Omura and R. Sato, “A New Cytochrome in Liver Microsomes,” Journal of Biological Chemistry, Vol. 237, No. 4, 1962, p. 1375.
- R. W. Estabrook, O. Rosenthal and D. Y. Cooper “Light Reversible Carbon Monoxide Inhibition of Steroid C21- Hydroxylase System of Adrenal Cortex,” Biochemische Zeitschrift, Vol. 338, 1963, p. 741.
- B. Meunier, S. P. de Visser and S. Shaik, “Mechanism of Oxidation Reactions Catalyzed by Cytochrome P-450 Enzymes,” Chemical Reviews, Vol. 104, No. 9, 2004, pp. 3947-3980. doi:10.1021/cr020443g
- B. Meunier, “Metalloporphyrins as Versatile Catalysts for Oxidation Reactions and Oxidative DNA Cleavage,” Chemical Reviews, Vol. 92, No. 6, 1992, 1411-1456. doi:10.1021/cr00014a008
- T. L. Poulos, “Peroxidase and Cytochrome P-450 Structures,” Academic Press, San Diego, 2000.
- S. Shaik, D. Kumar, S. P. de Visser, A. Altun and W. Thiel, “Theoretical Perspective on the Structure and Mechanism of Cytochrome P-450 Enzymes,” Chemical Reviews, Vol. 105, No. 6, 2005, pp. 2279-2328. doi:10.1021/cr030722j
- I. G. Denisov, T. M. Makris, S. G. Sligar and I. Schlichting, “Structure and Chemistry of Cytochrome P450,” Chemical Reviews, Vol. 105, 2005, pp. 2253-2277. doi:10.1021/cr0307143
- P. Hlavica, “Models and Mechanisms of O-O Bond Activation by Cytochrome P450-A Critical Assessment of the Potential Role of Multiple Active Intermediates in Oxidative Catalysis,” European Journal of Biochemistry, Vol. 271, No. 22, 2004, pp. 4335-4360. doi:10.1111/j.1432-1033.2004.04380.x
- D. Mansuy, “A Brief History of the Contribution of Metalloporphyrin Models to Cytochrome P-450 Chemistry and Oxidation Catalysis,” Comptes Rendus Chimie, Vol. 10, No. 4-5, 2007, pp. 392-413. doi:10.1016/j.crci.2006.11.001
- R. J. Nick, G. B. Ray, K. M. Fish, T. G. Spiro and J. T. Groves, “Evidence for a Weak Mn = O Bond and a Nonporphyrin Radical in Manganese-Substituted Horseradish-Peroxidase Compound-I,” Journal of the American Chemical Society, Vol. 113, No. 5, 1991, pp. 1838-1840. doi:10.1021/ja00005a062
- W. Nam, S. Park, I. K. Lim, M. H. Lim, J. Hong and J. Kim, “First Direct Evidence for Stereospecific Olefin Epoxidation and Alkane Hydroxylation by an Oxoiron(IV) Porphyrin Complex,” Journal of the American Chemical Society, Vol. 125, No. 48, 2003, pp. 14674-14675. doi:10.1021/ja0368204
- I. L. V. Rosa, C. M. C. P. Manso, O. A. Serra and Y. Iamamoto, “Biomimetical Catalytic Activity of Iron(III) Porphyrins Encapsulated in the Zeolite X,” Journal of Molecular Catalysis A—Chemical, Vol. 160, No. 2, 2000, pp. 199-208. doi:10.1016/S1381-1169(00)00214-4
- F. C. Skrobot, I. L. V. Rosa, A. P. A. Marques, P. R. Martins, J. Rocha, A. A. Valente and Y. Iamamoto, “Asymmetric Cationic Methyl Pyridyl and Pentafluorophenyl Porphyrin Encapsulated in zeolites: A Cytochrome P-450 model,” Journal of Molecular Catalysis A—Chemical, Vol. 237, No. 1-2, 2005, pp. 86-92. doi:10.1016/j.molcata.2005.05.001
- F. Bedioui, “Zeolite-Encapsulated and Clay-Intercalated Metal Porphyrin, Phthalocyanine and Schiff-Base Complexes as Models for Biomimetic Oxidation Catalysts— An Overview,” Coordination Chemistry Reviews, Vol. 144, 1995, pp. 39-68. doi:10.1016/0010-8545(94)08000-H
- G. Simonneaux, P. L. Maux, Y. Ferrand and J. RaultBerthelot, “Asymmetric Heterogeneous Catalysis by Metalloporphyrins,” Coordination Chemistry Reviews, Vol. 250, No. 17-18, 2006, pp. 2212-2221. doi:10.1016/j.ccr.2006.01.014
- Z. Li, C. G. Xia and X. M. Zhang, “Preparation and Catalysis of DMY and MCM-41 Encapsulated Cationic Mn(III)-Porphyrin Complex,” Journal of Molecular Catalysis A—Chemical, Vol. 185, No. 1-2, 2002, pp. 47-56. doi:10.1016/S1381-1169(02)00120-6
- P. S. Traylor, D. Dolphin and T. G. Traylor, “Sterically Protected Hemins with Electronegative Substituents— Efficient Catalysts for Hydroxylation and Epoxidation,” Journal of the Chemical Society-Chemical Communications, Vol. 5, 1984, pp. 279-280. doi:10.1039/c39840000279
- D. Mansuy and P. Battioni, “Metalloporphyrins in Catalytic Oxidations,” Marcel Dekker, New York, 1994.
- J. F. Bartoli, K. L. Barch, M. Palacio, P. Battioni, D. Mansuy, “Iron Polynitroporphyrins Bearing up to Eight Betanitro Groups as Interesting New Catalysts for H2O2-Dependent Hydrocarbon Oxidation: Unusual Regioselectivity in Hydroxylation of Alkoxybenzenes,” Chemical Communications, Vol. 18, No. 2, 2001, pp. 1718-1719. doi:10.1039/b105101f
- S. Evans and J. R. L. Smith, “The Oxidation of Ethylbenzene and other Alkylaromatics by Dioxygen Catalysed by Iron(III) tetrakis(pentafluorophenyl)porphyrin and Related Iron Porphyrins,” Journal of the Chemical SocietyPerkin Transactions 2, Vol. 7, 2000, pp. 1541-1551. doi:10.1039/b000967i
- S. H. Wang, B. S. Mandimutsira, R. Todd, B. Ramdhanie, J. P. Fox and D. P. Goldberg, “Catalytic Sulfoxidation and Epoxidation with a Mn(III) Triazacorrole: Evidence for a Third Oxidant in High-Valent Porphyrinoid Oxidations,” Journal of the American Chemical Society, Vol. 126, No. 1, 2004, pp. 18-19. doi:10.1021/ja038951a
- M. S. M. Moreira, P. R. Martins, R. B. Curi, O. R. Nascimento and Y. Iamamoto, “Iron Porphyrins Immobilized on Silica Surface and Encapsulated in Silica Matrix: A Comparison of Their Catalytic Activity in Hydrocarbon Oxidation,” Journal of Molecular Catalysis A—Chemical, Vol. 233, No. 1-2, 2005, pp. 73-81. doi:10.1016/j.molcata.2005.01.045
- K. J. Ciuffi, H. C. Sacco, J. B. Valim, C. M. C. P. Manso, O. A. Serra, O. R. Nascimento, E. A. Vidoto and Y. Iamamoto, “Polymeric Organic-Inorganic Hybrid Material Containing Iron(III) Porphyrin Using Sol-Gel Process,” Journal of Non-Crystalline Solids, Vol. 247, 1999, pp. 146- 152. doi:10.1016/S0022-3093(99)00053-8
- K. J. Ciuffi, H. C. Sacco, J. C. Biazzotto, E. A. Vidoto, O. R. Nascimento, C. A. P. Leite, O. A. Serra and Y. Iamamoto, “Synthesis of Fluorinated Metalloporphyrinosilica Imprinted with Templates through Sol-gel Process,” Journal of Non-Crystalline Solids, Vol. 273, No. 1-3, 2000, pp. 100-108. doi:10.1016/S0022-3093(00)00149-6
- C. M. C. P. Manso, E. A. Vidoto, F. S. Vinhado, H. C. Sacco, P. R. Martins, K. J. Ciuffi, A. G. Ferreira, J. R. Lindsay-Smith, O. R. Nascimento and Y. Iamamoto, “Characterization and Catalytic Activity of Iron(III) mono(4-Nmethyl pyridyl)-tris(halophenyl) Porphyrins in Homogeneous and Heterogeneous Systems,” Journal of Molecular Catalysis A—Chemical, Vol. 150, No. 1-2, 1999, pp. 251-266. doi:10.1016/S1381-1169(99)00216-2
- F. S. Vinhado, P. R. Martins, A. P. Masson, D. G. Abreu, E. A. Vidoto, Y. Iamamoto and O. R. Nascimento, “Supported Iron(III)porphyrins Pentafluorophenyl-derivatives as Catalysts in Epoxidation Reactions by H2O2: The Role of the Silica-support and Sulfonatophenyl Residues in the Activation of the Peroxidic Bond,” Journal of Molecular Catalysis A—Chemical, Vol. 188, No. 1-2, 2002, pp. 141- 151. doi:10.1016/S1381-1169(02)00233-9
- F. S. Vinhado, C. M. C. P. Manso, H. C. Sacco and Y. Iamamoto, “Cationic Manganese(III) Porphyrins Bound to a Novel Bis-Functionalised Silica as Catalysts for Hydrocarbons Oxygenation by Iodosylbenzene and Hydrogen Peroxide,” Journal of Molecular Catalysis A—Chemical, Vol. 174, No. 1-2, 2001, pp. 279-288. doi:10.1016/S1381-1169(01)00200-X
- A. L. Faria, C. Airoldi, F. G. Doro, M. G. Fonseca and M. D. Assis, “Anchored Ironporphyrins—The Role of TalcAminofunctionalyzed Phyllosilicates in the Catalysis of Oxidation of Alkanes and Alkenes,” Applied Catalysis A-General, Vol. 268, No. 1-2, 2004, pp. 217-226. doi:10.1016/j.apcata.2004.03.035
- A. A. Guedes, J. R. L. Smith, O. R. Nascimento, D. F. C. Guedes and M. D. Assis, “Catalytic Activity of Halogenated Iron Porphyrins in Alkene and Alkane Oxidations by Iodosylbenzene and Hydrogen Peroxide,” Journal of the Brazilian Chemical Society, Vol. 16, No. 4, 2005, pp. 835-843.doi:10.1590/S0103-50532005000500024
- M. A. Schiavon, L. S. Iwamoto, A. G. Ferreira, Y. Iamamoto, M. V. B. Zanoni and M. D. Assis, “Synthesis and Characterization of a Novel Series of Meso (Nitrophenyl) and Meso (CarboxyPhenyl) Substituted porphyrins,” Journal of the Brazilian Chemical Society, Vol. 11, No. 5, 2000, pp. 458-466. doi:10.1590/S0103-50532000000500005
- A. E. Shilov, “Chemical-Models of Metallo-Enzymes,” Journal of Molecular Catalysis, Vol. 47, No. 2-3, 1988, pp. 351-362. doi:10.1016/0304-5102(88)85060-0
- B. R. Cook, T. J. Reinert and K. S. Suslick, “Shape Selective Alkane Hydroxylation by Metalloporphyrin Catalysts,” Journal of the American Chemical Society, Vol. 108, No. 23, 1986, pp. 7281-7286. doi:10.1021/ja00283a024
- M. J. Nappa and C. A. Tolman, “Steric and Electronic Control of Iron Porphyrin Catalyzed Hydrocarbon Oxidations.” Inorganic Chemistry, Vol. 24, No. 26, 1985, pp. 4711-4719. doi:10.1021/ic00220a056
- Z. Gross and L. Simkhovich, “Hydroxylation of Simple Alkanes by Iodosylbenzene in Catalyzed more Efficiently by Second than by Third Generation Iron(III) Porphyrins,” Tetrahedron Letters, Vol. 39, No. 44, 1998, pp. 8171-8174. doi:10.1016/S0040-4039(98)01820-6
- A. Corma, “State of the Art and Future Challenges of Zeolites as Catalysts,” Journal of Catalysis, Vol. 216, No. 1-2, pp. 298-312. doi:10.1016/S0021-9517(02)00132-X
- Y. Iamamoto and K. J. Ciuffi, “Characterization and Catalytic Activity of 2,6-Dichlorophenyl Substituted Iron(III) Porphyrin Supported on Silica Gel and Imidazole Propyl Gel,” Journal of Molecular Catalysis A—Chemical, Vol. 116, No. 3, 1997, pp. 405-420. doi:10.1016/S1381-1169(96)00342-1
- R. F. Parton, G. J. Peere, P. E. Neysa, P. A. Jacobs, R. Claessens and G. V. Baron, “Cyclohexane Oxidation with Tertiary-Butylhydroperoxide Catalyzed by Iron-Phthalocyanines Homogeneously and Occluded in Y Zeolite,” Journal of Molecular Catalysis A–Chemical, Vol. 113, No. 3, 1996, pp. 445-454. doi:10.1016/S1381-1169(96)00111-2
- B. Z. Zhan and X. Y. Li, “A Novel ‘build-bottlearound-ship’ Method to Encapsulate Metalloporphyrins in Zeolite-Y. An Efficient Biomimetic Catalyst,” Chemical Communications, Vol. 1998, No. 3, 1998, pp. 349-350. doi:10.1039/a706030k
- F. C. Skrobot, “Synthesis and Haracterization of Manganese and Iron Porphyrins Encapsulated in Zeolites: Catalytics Studies in the Homogeneous and Heterogeneous Systems,” Thesis, University of São Paulo, São Carlos, 2004.
- F. C. Skrobot, A. A. Valente, G. Nevesa, I. Rosa, J. Rocha and J. A. S. Cavaleiro, “Monoterpenes Oxidation in the Presence of Y Zeolite-Entrapped Manganese(III) Tetra (4-N-benzylpyridyl)porphyrin,” Journal of molecular catalysis A–Chemical, Vol. 201, No. 1-2, 2003, pp. 211-222. doi:10.1016/S1381-1169(03)00181-X
- T. A. Khan and J. A. Hriljac, “Hydrothermal Synthesis of Microporous Materials with the Direct Incorporation of Porphyrin Molecules,” Inorganica Chimica Acta, Vol. 294, No. 2, 1999, pp. 179-182. doi:10.1016/S0020-1693(99)00247-9
- C. V. McDaniel and P. K. Maher, “Zeolite Chemistry and Catalysis,” ACS, Washington DC, 1976.
- K. J. Balkus Jr., A. G. Gabrielov, S. L. Bell, F. Bedioui, L. Roue and J. Devynck, “Zeolite Encapsulated Cobalt(ii) and Copper(ii) Perfluorophthalocyanines-Synthesis and Characterization,” Inorganic Chemistry, Vol. 33, No. 1, 1994, pp. 67-72. doi:10.1021/ic00079a013
- W. Zhang, P. G. Smirniotis, M. Gangoda and R. N. Bose, “Bronsted and Lewis Acid sites in Dealuminated ZSM-12 and Beta Zeolites Characterized by NH3-STPD, FT-IR, and MAS NMR Spectroscopy,” Journal of Physical Chemistry B, Vol. 104, No. 17, 2000, pp. 4122-4129. doi:10.1021/jp993072p
- J. L. McLain, J. Lee and J. T. Groves, “Biomimetic Oxidations Catalyzed by Transition Metal Complexes,” Imperial College Press, London, 2000, p. 91. doi:10.1142/9781848160699_0003
- J. T. Groves, W. J. Kruper and R. C. Haushalter, “Hydrocarbon Oxidations with Oxometalloporphinate. Isolation and Reactions of a (Porphinato)Manganese(v) Complex,” Journal of the American Chemical Society, Vol. 102, No. 20, 1980, pp. 6375-6377. doi:10.1021/ja00540a050
- G. S. Nunes, I. Mayer, H. E. Toma, K. Araki, “Kinetics and Mechanism of Cyclohexane Oxidation Catalyzed by Supramolecular Manganese(III) Porphyrins,” Journal of Catalysis, Vol. 236 No.1, 2005, pp. 55-61. doi:10.1016/j.jcat.2005.09.003
- J. R. Lindsay-Smith, Y. Iamamoto and F. S. Vinhado, “Oxidation of Alkanes by Iodosylbenzene (PhIO) Catalysed by Supported Mn(III) Porphyrins: Activity and Mechanism,” Journal of Molecular Catalysis A—Chemical, Vol. 252, No. 1-2, 2006, pp. 23-30. doi:10.1016/j.molcata.2006.01.064
- D. H. R. Barton and D. Doller, “The Selective Functionalization of Saturated Hydrocarbons—Gif Chemistry,” Accounts of Chemical Research, Vol. 25, No. 11, 1992, pp. 504-512. doi:10.1021/ar00023a004
- J. T. Groves, T. E. Nemo and R. S. Myers, “Hydroxylation and Epoxidation Catalyzed by Iron-Porphine Complexes-Oxygen-Transfer from Iodosylbenzene,” Journal of the American Chemical Society, Vol. 101, No. 4, 1979, pp. 1032-1033. doi:10.1021/ja00498a040
NOTES
*Corresponding author.