Pharmacology & Pharmacy
Vol.5 No.4(2014), Article ID:44660,8 pages DOI:10.4236/pp.2014.54044
Use of Human Embryonic Stem Cell-Derived Cardiomyocyte Clusters to Assess Potential for Chronic Treatment with Compounds to Cause QT Prolongation
Taro Hihara1, Kazuto Yamazaki2*, Tomohiko Taniguchi3, Takashi Yoshinaga3, Masashi Ito2, Kohei Sawada3
1Biomarkers and Personalized Medicine CFU, Eisai Product Creation Systems, Eisai Co., Ltd., Tsukuba, Japan
2Next Generation Systems CFU, Eisai Product Creation Systems, Eisai Co., Ltd., Tsukuba, Japan
3Biopharmaceutical Assessment CFU, Eisai Product Creation Systems, Eisai Co., Ltd., Tsukuba, Japan
Email: *k5-yamazaki@hhc.eisai.co.jp
Copyright © 2014 by authors and Scientific Research Publishing Inc.
This work is licensed under the Creative Commons Attribution International License (CC BY).
http://creativecommons.org/licenses/by/4.0/


Received 6 February 2014; revised 16 March 2014; accepted 27 March 2014
Abstract
Drug-induced QT prolongation is a serious clinical issue in developing novel drug candidates and marketing drugs. A major cause of QT prolongation is direct inhibition of human ether-à-go-go-related gene (hERG) channels. Reduction in repolarization-related channel expression levels on plasma membranes is another mechanism that induces QT prolongation. Recently, we established a system for assessing the risk of QT prolongation by using human embryonic stem cell-derived cardiomyocyte clusters (hES-CMCs) in which the field potential duration (FPD) or corrected FPD (FPDc) was measured as an indicator of drug-induced QT interval prolongation. Here, we examined whether this system was able to detect FPDc prolongation caused by pentamidine or probucol, both of which can induce QT prolongation after long-term treatment. hES-CMCs were treated with pentamidine or probucol, and the FPDc of the same clusters was measured 10 min, 4 h, and 24 h after the start of treatment. Concentration-dependent FPDc prolongation was observed at 24 h, but not at 10 min, with pentamidine or probucol treatment. These results suggest that the hES-CMC-based assessment system can be used to detect both acute (at 10 min) and delayed (at 24 h) QT prolongation risk on the same platform by simple alteration of the extended culture period.
Keywords
Human Embryonic Stem Cell-Derived Cardiomyocytes; Field Potential Duration; QT Prolongation; Chronic Treatment; Risk Assessment

1. Introduction
QT prolongation, torsades de pointes (TdP), and arrhythmias are serious problems in the development of novel drug candidates and the marketing of drugs. Inhibition of human ether-à-go-go-related gene (hERG) channels, which conduct IKr currents, is the mechanism most frequently involved in these episodes and is the most important target of safety assessment in drug development. Pharmaceutical companies have therefore been using hERG channel-overexpressing cells—that is, hERG assays—to screen drug candidate compounds for hERG channel inhibition. Besides hERG channel inhibition, loss of function of IKs channels, which are composed of KCNQ1 (KvLQT1) and KCNE1, is a well-known trigger of QT prolongation [1] . There are two main mechanisms by which these QT-related channels are involved in drug-induced QT prolongation: one is by direct action on the channels (e.g. inhibition of hERG channels through binding of E-4031 to the channels [2] ), and the other is indirect (e.g. inhibition of hERG channel trafficking by pentamidine [3] or reduction of surface membrane expression of KCNQ1 by probucol [4] ). The latter mechanisms cause QT prolongation in chronic drug treatment.
We recently established an in vitro system that uses human embryonic stem cell-derived cardiomyocyte clusters (hES-CMCs) to assess the potential of compounds to influence QT intervals [5] . In this hES-CMC system, we record the field potential duration (FPD) (i.e. the QT-like interval) of clusters treated with a compound; we then calculate the corrected FPD (FPDc) and examine whether or not the compound prolongs FPDc. In our previous study, we evaluated E-4031, cisapride, and dl-sotalol as reference hERG channel blockers, and these compounds manifested acute FPDc prolongation [5] . However, there have been few reports assessing compounds that can cause QT prolongation through the indirect mechanisms above-mentioned by using FPDc as a parameter for QT prolongation. Therefore, in the next stage of our research, we set up a platform to assess the potential for QT prolongation by indirect mechanisms. Here, we report the effects of pentamidine and probucol on FPDc of hES-CMCs in culture for a long period. We show the usefulness of the system for evaluating compounds with direct or indirect effects on repolarization-related channels. We also demonstrate the possible interaction of multiple repolarization-related channels.
2. Materials and Methods
2.1. Chemicals and Reagents
Pentamidine isethionate and probucol were purchased from Wako Pure Chemical Industries, Ltd. (Osaka, Japan). Cisapride was obtained from Sigma-Aldrich (St. Louis, MO). Pentamidine and cisapride were dissolved in dimethyl sulfoxide (DMSO; Sigma-Aldrich) at 10 mM as stock solutions. Probucol was dissolved in 99.5% ethanol (Wako Pure Chemical Industries) at 10 mM as a stock solution.
Dulbecco’s modified Eagle’s medium (DMEM) was purchased from Sigma-Aldrich. Non-essential amino acids (100×, NEAA), 100× penicillin/streptomycin solution, and β-mercaptoethanol (55 mM) were purchased from Invitrogen (Carlsbad, CA). Fetal bovine serum (FBS) was obtained from Equitech-Bio (Kerrville, TX).
2.2. Use of 24-h hES-CMC Treatment to Assess the Potential of Compounds to Influence QT Interval
The basic method has been described in detail by Yamazaki et al. [5] . hES-CMCs (hES-CMC002) were obtained from Cellectis AB (formerly Cellartis AB; Göteborg, Sweden) and incubated in DMEM supplemented with 1× NEAA, 1× penicillin/streptomycin, 0.1 mM β-mercaptoethanol, and 20% heat-inactivated FBS at 37˚C in a 5% CO2 incubator. The clusters were adhered to the electrodes of the probes (MED-P515A; Alpha MED Sciences, Osaka, Japan) of a MED64 System, a multielectrode device for recording field potentials. The preparations were placed on a hot plate at 37˚C and covered with a lid, through which aeration with 95% O2/5% CO2 gas was performed. Initially the clusters were treated with cisapride (a hERG channel blocker) at 100 nM, and their FPDs, inter-spike intervals (ISIs), and beat frequencies were recorded with the MED64 System with its own software (Mobius QT; Alpha MED Sciences). The FPDs with the vehicle and cisapride treatments were measured for 10 min. The last 30 FPDs and ISIs of the 10 min-recordings were extracted for analysis. FPDc was calculated according to Fridericia’s formula: FPDc = FPD/ISI1/3. hES-CMCs showing 5% to 20% FPDc prolongation upon treatment with 100 nM cisapride were selected for the next examination. The washout period was 7 days.
Before the addition of pentamidine or probucol, clusters were treated with vehicle (0.1% DMSO in the case of the pentamidine groups or 0.1% ethanol for the probucol groups) to obtain pre-values (pre-treatment). hESCMCs were then treated with pentamidine (1, 3, or 10 µM), probucol (0.1, 0.3, 1, 3, or 10 µM), or vehicle (DMSO for the pentamidine groups or ethanol for the probucol groups; final concentrations were 0.2% for both). In this assay, we cultured the clusters in DMEM supplemented with 20% heat-inactivated FBS, 1× penicillin/streptomycin, 1× NEAA, 0.1 mM β-mercaptoethanol, 50 mM NaCl, and 15 mM NaHCO3, adjusted to pH 7.2 with 1 N NaOH. FPDs, ISIs, and beat frequencies were recorded at pre-treatment and 10 min, 4 h, and 24 h after the addition of each compound. The assessment flow is depicted in Figure 1.
2.3. Confirmation of Responsiveness of hES-CMCs to Cisapride after 24-h Vehicle Treatment
To exclude the effects of 24-h vehicle treatment (0.2% DMSO or 0.2% ethanol) on hERG channel function, we treated the 24-h vehicle-treated clusters with cisapride at 100 nM for 10 min to confirm that they showed FPDc prolongation through the hERG channel-blocking action of this drug.
2.4. Statistical Analysis
All values are expressed as means ± S.E.M. A two-tailed paired Student’s t-test was conducted to analyze changes in FPDc. p values less than 0.05 were considered significant.
3. Results and Discussion
In the vehicle-treated groups (0.2% DMSO for the pentamidine-treated groups, or 0.2% ethanol for the probucol-treated groups) there were no significant changes in FPDc at the 10-min or 4- and 24-h treatment points (Tables 1 and 2). Clusters treated with 0.2% DMSO or 0.2% ethanol for 24 h responded to cisapride at 100 nM: at the 24-h vehicle treatment point, FPDc was prolonged by 22.1 ± 4% in the 0.2% DMSO-treated clusters (p
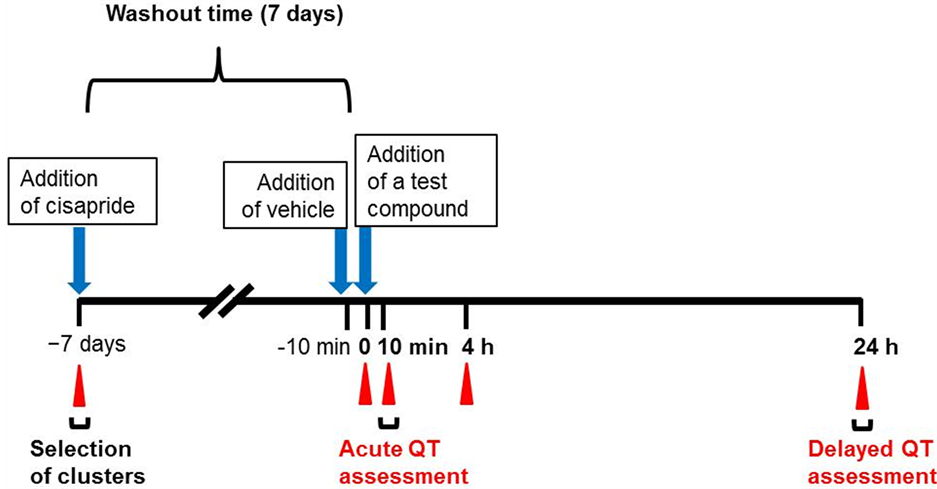
Figure 1. Scheme for using human embryonic stem cell-derived cardiomyocyte clusters (hES-CMCs) to assess the potential of compounds to cause acute or delayed QT prolongation. hES-CMCs were treated with cisapride at 100 nM and their field potential duration (FPD), inter-spike intervals (ISIs), and beat frequencies were measured to select clusters appropriate for the assessment. Clusters with 5 to 20% corrected FPD (FPDc) were chosen. After 7 days’ washout, the clusters were treated with pentamidine (1, 3, or 10 µM), probucol (0.1, 0.3, 1, 3, or 10 µM), or vehicle (dimethyl sulfoxide in the case of pentamidine, or ethanol in the case of probucol). FPD, ISIs, and beat frequencies were measured at pre-treatment and 10 min, 4 h, and 24 h after addition of the compound or vehicle. Blue arrows indicate the time at which a compound or vehicle was added, and red triangles indicate the times at which FPD was measured.

Table 1. Effects of exposure to pentamidine for up to 24 h on the corrected field potential duration (FPDc) of human embryonic stem cell-derived cardiomyocyte clusters. FPDc was calculated before, and 10 min, 4 h, and 24 h after, the addition of pentamidine.
Field potential duration (FPD) with vehicle (0.2% dimethyl sulfoxide) and with each concentration of pentamidine was measured for 10 min. The last 30 FPDs and inter-spike intervals (ISIs) of the 10 min-recordings were extracted for analysis. FPDc was calculated according to Fridericia’s formula: FPDc = FPD/ISI1/3. Values are expressed as means ± S.E.M. #, p < 0.10; *, p < 0.05.
Table 2. Effects of exposure to probucol for up to 24 h on the corrected field potential duration (FPDc) of human embryonic stem cell-derived cardiomyocyte clusters. FPDc was calculated before, and 10 min, 4 h, and 24 h after, the addition of probucol.
Field potential duration (FPD) with vehicle (0.2% ethanol) and with each concentration of probucol was measured for 10 min. The last 30 FPDs and inter-spike intervals (ISIs) of the 10 min-recordings were extracted for analysis. FPDc was calculated according to Fridericia’s formula: FPDc = FPD/ISI1/3. Values are expressed as means ± S.E.M. *, p < 0.05; **, p < 0.01.
< 0.05, n = 4) and by 21.1 ± 3.5% in the 0.2% ethanol-treated clusters (p < 0.01, n = 4). These findings suggested that neither 24-h 0.2% DMSO nor 0.2% ethanol treatment impeded hERG channel function.
Pentamidine treatment did not cause significant FPDc prolongation at 10 min, except with the 3-µM treatment (p < 0.05, actual values) (Table 1). However, this prolongation corresponded to only 2.1% above the pre-FPDc value. Because we regarded >5% prolongation to be of toxicological significance, this degree of prolongation was not considered meaningful. No significant prolongation was observed at 4 h, but a trend in prolongation was seen in the 3 µM group (p = 0.0891, actual values). Clear concentration-dependent FPDc prolongation was detected at 24 h: a trend in prolongation was found with the 1- and 3-µM treatments (p = 0.0755 and p = 0.0753, respectively): two of three clusters showed >10.0% FPDc prolongation in the 1-µM group, and all four clusters showed >8.0% prolongation in the 3-µM group. Significant, prominent prolongation of FPDc was triggered by 10-µM pentamidine treatment (p < 0.05), which gave a 62.0% increase over the pre-value.
Pentamidine has been used to treat Pneumocystis jirovecii pneumonia in patients infected with human immunodeficiency virus [6] . This agent has also been used to treat a variety of parasitic diseases, including trypanosomiasis and leishmaniasis [7] [8] . In clinical use, pentamidine is administered via daily intramuscular injection or intravenous (i.v.) infusion at 4 mg/kg body weight. The plasma maximum concentration (Cmax) has been reported as 612 ng/mL (1.8 µM total or 0.54 µM free) within 60 min of 4 mg/kg i.v. injection [9] . QT prolongation, TdP, and tachycardia are well-reported adverse events in treatment with this drug [10] -[13] . Most notably, the majority of TdP episodes have occurred in patients undergoing prolonged pentamidine treatment, instead of immediately following administration [10] [11] [13] -[16] : Wharton et al. reported two cases in which patients developed QT prolongation 20 and 13 days after the start of pentamidine treatment at 4 mg/kg daily [10] .
Kuryshev et al. reported that overnight (16 to 20 h) treatment with pentamidine reduced hERG currents and inhibited trafficking and maturation (full glycosylation) of hERG channels in hERG channel-overexpressing HEK (human embryonic kidney) 293 cells in a concentration-dependent manner; the IC50 values were 5 to 8 µM [17] . Pentamidine at 10 µM prolonged the action potential duration at 90% repolarization (APD90) in isolated guinea pig ventricular myocytes incubated overnight (24 h). Cordes et al. found that pentamidine inhibited hERG currents directly (IC50 value, 252 µM) in HEK293 cells stably overexpressing hERG channels; this is far greater than the relevant maximum clinical concentration [18] . They found reduced hERG currents, reduced quantity of hERG polypeptides, and reduced density of hERG proteins on the surface membranes of hERGoverexpressing HEK293 cells treated with pentamidine at 1 or 10 µM for 48 h. Interestingly, Yokoyama et al. showed that acute single i.v. administration of a clinically relevant dose (Cmax values were estimated to be about 1.7 to 5.0 µM) of pentamidine prolonged corrected QT (QTc) intervals in halothane-anesthetized dogs [19] . They speculated that inhibition of IKr channel trafficking by pentamidine was caused more acutely than previously expected. But our in vitro study did not indicate clear FPDc prolongation 10 min after the addition of pentamidine at 1, 3, and 10 µM in hES-CMCs, suggesting that a possibility that there is a species difference of sensitivity of hERG channel trafficking by pentamidine.
We observed a trend of FPDc prolongation in hES-CMCs at 24 h upon treatment with pentamidine at 1 (8.2%) or 3 (26.1%) µM. Considering that the plasma Cmax value of pentamidine is 1.8 µM (total) in men [9] , our results at 1 and 3 µM pentamidine seem reasonable in explaining clinical QT prolongation. On the other hand, we did not expect that pentamidine at 10 µM would prolong FPDc at 24 h to a greater extent (62%) than did hERG channel blockage by cisapride at 100 nM (~20%). (The calculated IC50 value of cisapride against hERG currents is 13.9 nM in hERG channel-overexpressing cells [20] ). This unexpectedly long prolongation may have been caused by (as yet unknown) mechanisms other than inhibition of hERG trafficking by pentamidine.
With probucol treatment of hES-CMCs there were no significant changes in FPDc at 10 min (Table 2). At 4 h there was a significant prolongation of FPDc (p < 0.05 and p < 0.01 for the 3- and 10-µM treatments, respectively, compared with the pre-treatment values), although these prolongations were only 4.2% and 3.1%, respectively, above the pre-values. Significant and >5% FPDc prolongation was observed at 24 h with 0.3- (p < 0.05), 1- (p < 0.05), 3- (p < 0.01), and 10- (p < 0.05) µM probucol treatment.
Probucol is a cholesterol-lowering drug used to treat hyperlipidemia, including familial hypercholesterolemia and xanthoma [21] . Jeon et al. studied the pharmacokinetics of probucol in healthy volunteers who received probucol orally at 250 mg once daily (q.d.), 500 mg q.d., or 250 mg twice a day (b.i.d.) for 14 days [22] . Plasma concentrations reached ~4 µg/mL (7.7 µM) in all groups on day 1 (24 h) and plateaued as follows on day 13: ~6 µg/mL (11.6 µM) in the 250-mg q.d. group, ~8 µg/mL (15.5 µM) in the 500-mg q.d. group, and ~10 µg/mL (19.3 µM) in the 250-mg b.i.d. group. Probucol can cause QT prolongation [23] , and TdP and arrhythmia have been reported in some patients [24] -[26] : Tamura et al. described the case of a hypercholesterolemic female patient receiving 500 mg b.i.d. who experienced two episodes of syncope [26] . Her QTc intervals became prolonged 12 weeks after the start of probucol treatment and decreased to about the baseline value by 6 weeks after its discontinuation.
Guo et al. reported that IKr currents were reduced in rat neonatal ventricular myocytes (IC50 = 20.6 µM) and in hERG channel-overexpressing HEK293 cells (IC50 = 10.6 µM) by 48-h probucol exposure; this reduction was not observed in acute treatment [27] . APD90 prolongation was observed in rat neonatal cardiomyocytes treated with 30 µM probucol for 24 h. Probucol treatment at 100 µM for 48 h significantly reduced the mature plasma membrane forms of hERG channel protein in hERG channel-overexpressing cells and rat neonatal cardiomyocytes. Furthermore, Guo et al. found that probucol treatment reduced hERG channel expression by accelerating caveolin-1 turnover [28] . More recently, Taniguchi et al. investigated the mechanisms of delayed QT prolongation of probucol by using both KCNQ1- and KCNE1-co-overexpressing CHO (Chinese hamster ovary)-K1 cells [4] . Using these cells, the acute inhibitory effects of probucol at concentrations of 0.003 to 10 µM on IKs currents were examined. A significant reduction in IKs currents was found at 0.03 and 0.1 µM, but this reduction was not concentration-dependent. In contrast, probucol significantly reduced the IKs currents at 0.3 to 1 µM in 2- and 8-h treatments, and at 0.03 to 1 µM in 8- and 24-h treatments. Western blot analysis revealed that the abundance of a multimeric complex of KCNQ1 proteins was diminished after 24 h of treatment with probucol at ≥0.03 µM. Taniguchi et al. showed that probucol treatment at 1 to 30 µM for 24 h also inhibited hERG currents in hERG channel-overexpressing CHO-K1 cells, but the inhibitory effect was not concentration-dependent and was very slight (~20% at maximum) [4] . They therefore concluded that probucol inhibited IKs currents more specifically than IKr currents.
In consideration of the fact that the clinical concentrations of probucol (~8 to 20 µM) are much higher than those that inhibit IKs currents (0.03 to 1 µM), QT prolongation and TdP resulting from probucol may be caused by reduced plasma membrane expression of both hERG and IKs channels. Our study showed that FPDc was significantly prolonged at ≥0.3 µM with 24-h probucol treatment. Taking these findings together, we can speculate that FPDc prolongation at low concentrations (≤~1 µM) of probucol is due to the inhibition of IKs currents and that prolongation at >1 µM may be a result of combined inhibition of both IKs and IKr currents. As formation of a macrocomplex of hERG and KCNQ1 channels on plasma membranes has been reported by Guo et al. [29] , it is likely that the effects of a compound inhibiting both hERG and IKs channels on QT intervals are additive or synergistic.
4. Conclusion
We observed FPDc prolongation of hES-CMCs after 24 h of treatment with pentamidine and probucol, both of which prolong QTc intervals in chronic exposure clinically. Application of this experimental protocol to FPDc measurement at 10 min and 24 h should enable us to assess the potential for acute and delayed occurrence of QT prolongation in the same clusters in response to drug candidates, thus saving time and reducing costs.
References
- Hedley, P.L., Jørgensen, P., Schlamowitz, S., Wangari, R., Moolman-Smook, J., Brink, P.A., Kanters, J.K., Corfield, V.A. and Christiansen, M. (2009) The Genetic Basis of Long QT and Short QT Syndromes: A Mutation Update. Human Mutation, 30, 1486-1511. http://dx.doi.org/10.1002/humu.21106
- Finlayson, K., Turnbull, L., January, C.T., Sharkey, J. and Kelly, J.S. (2001) [3H]Dofetilide Binding to HERG Transfected Membranes: A Potential High Throughput Preclinical Screen. European Journal of Pharmacology, 430, 147- 148. http://dx.doi.org/10.1016/S0014-2999(01)01362-0
- Dennis, A., Wang, L., Wan, X. and Ficker, E. (2007) hERG Channel Trafficking: Novel Targets in Drug-Induced Long QT Syndrome. Biochemical Society Transactions, 35, 1060-1063. http://dx.doi.org/10.1042/BST0351060
- Taniguchi, T., Uesugi, M., Arai, T., Yoshinaga, T., Miyamoto, N. and Sawada, K. (2012) Chronic Probucol Treatment Decreases the Slow Component of the Delayed-Rectifier Potassium Current in CHO Cells Transfected with KCNQ1 and KCNE1: A Novel Mechanism of QT Prolongation. Journal of Cardiovascular Pharmacology, 59, 377-386. http://dx.doi.org/10.1097/FJC.0b013e318245e0c5
- Yamazaki, K., Hihara, T., Taniguchi, T., Kohmura, N., Yoshinaga, T., Ito, M. and Sawada, K. (2012) A Novel Method of Selecting Human Embryonic Stem Cell-Derived Cardiomyocyte Clusters for Assessment of Potential to Influence QT Interval. Toxicology in Vitro, 26, 335-342. http://dx.doi.org/10.1016/j.tiv.2011.12.005
- Goa, K.L. and Campoli-Richards, D.M. (1987) Pentamidine Isethionate. A Review of Its Antiprotozoal Activity, Pharmacokinetic Properties and Therapeutic Use in Pneumocystis carinii Pneumonia. Drugs, 33, 242-258. http://dx.doi.org/10.2165/00003495-198733030-00002
- Nacher, M., Carme, B., Sainte Marie, D., Couppié, P., Clyti, E., Guibert, P. and Pradinaud, R. (2001) Influence of Clinical Presentation on the Efficacy of a Short Course of Pentamidine in the Treatment of Cutaneous Leishmaniasis in French Guiana. Annals of Tropical Medicine and Parasitology, 95, 331-336. http://dx.doi.org/10.1080/00034980120064355
- Burchmore, R.J., Ogbunude, P.O., Enanga, B. and Barrett, M.P. (2002) Chemotherapy of Human African Trypanosomiasis. Current Pharmaceutical Design, 8, 256-267. http://dx.doi.org/10.2174/1381612023396159
- Conte, Jr., J.E., Upton, R.A., Phelps, R.T., Wofsy, C.B., Zurlinden, E. and Lin, E.T. (1986) Use of a Specific and Sensitive Assay to Determine Pentamidine Pharmacokinetics in Patients with AIDS. The Journal of Infectious Diseases, 154, 923-929. http://dx.doi.org/10.1093/infdis/154.6.923
- Wharton, J.M., Demopulos, P.A. and Goldschlager, N. (1987) Torsade de Pointes during Administration of Pentamidine Isethionate. The American Journal of Medicine, 83, 571-576. http://dx.doi.org/10.1016/0002-9343(87)90774-1
- Bibler, M.R., Chou, T.C., Toltzis, R.J. and Wade, P.A. (1988) Recurrent Ventricular Tachycardia Due to Pentamidine-Induced Cardiotoxicity. Chest, 94, 1303-1306. http://dx.doi.org/10.1378/chest.94.6.1303
- Girgis, I., Gualberti, J., Langan, L., Malek, S., Mustaciuolo, V., Costantino, T. and McGinn, T.G. (1997) A Prospective Study of the Effect of IV Pentamidine Therapy on Ventricular Arrhythmias and QTc Prolongation in HIV-Infected Patients. Chest, 112, 646-653. http://dx.doi.org/10.1378/chest.112.3.646
- Kroll, C.R. and Gettes, L.S. (2002) T Wave Alternans and Torsades de Pointes after the Use of Intravenous Pentamidine. Journal of Cardiovascular Electrophysiology, 13, 936-938. http://dx.doi.org/10.1046/j.1540-8167.2002.00936.x
- Cortese, L.M., Gasser Jr., R.A., Bjornson, D.C., Dacey, M.J. and Oster, C.N. (1992) Prolonged Recurrence of Pentamidine-Induced Torsades de Pointes. The Annals of Pharmacotherapy, 26, 1365-1369.
- Eisenhauer, M.D., Eliasson, A.H., Taylor, A.J., Coyne Jr., P.E. and Wortham, D.C. (1994) Incidence of Cardiac Arrhythmias during Intravenous Pentamidine Therapy in HIV-Infected Patients. Chest, 105, 389-395. http://dx.doi.org/10.1378/chest.105.2.389
- Otsuka, M., Kanamori, H., Sasaki, S., Taguchi, J., Harano, H., Ogawa, K., Matsuzaki, M., Mohri, H., Okubo, T., Sumita, S. and Ochiai, H. (1997) Torsades de Pointes Complicating Pentamidine Therapy of Pneumocystis carinii Pneumonia in Acute Myelogenous Leukemia. Internal Medicine (Tokyo), 36, 705-708. http://dx.doi.org/10.2169/internalmedicine.36.705
- Kuryshev, Y.A., Ficker, E., Wang, L., Hawryluk, P., Dennis, A.T., Wible, B.A., Brown, A.M., Kang, J., Chen, X.-L., Sawamura, K., Reynolds, W. and Rampe, D. (2005) Pentamidine-Induced Long QT Syndrome and Block of hERG Trafficking. The Journal of Pharmacology and Experimental Therapeutics, 312, 316-323. http://dx.doi.org/10.1124/jpet.104.073692
- Cordes, J.S., Sun, Z., Lloyd, D.B., Bradley, J.A., Opsahl, A.C., Tengowski, M.W., Chen, X. and Zhou, J. (2005) Pentamidine Reduces hERG Expression to Prolong the QT Interval. British Journal of Pharmacology, 145, 15-23. http://dx.doi.org/10.1038/sj.bjp.0706140
- Yokoyama, H., Nakamura, Y., Iwasaki, H., Nagayama, Y., Hoshiai, K., Mitsumori, Y. and Sugiyama, A. (2009) Effects of Acute Intravenous Administration of Pentamidine, a Typical hERG-Trafficking Inhibitor, on the Cardiac Repolarization Process of Halothane-Anesthetized Dogs. Journal of Pharmacological Sciences, 110, 476-482. http://dx.doi.org/10.1254/jphs.09071FP
- Yamazaki, K., Hihara, T., Kato, H., Fukushima, T., Fukushima, K., Taniguchi, T., Yoshinaga, T., Miyamoto, N., Ito, M. and Sawada, K. (2014) Beat-to-Beat Variability in Field Potential Duration in Human Embryonic Stem Cell-Derived Cardiomyocyte Clusters for Assessment of Arrhythmogenic Risk, and a Case Study of Its Application. Pharmacology and Pharmacy, 5, 117-128. http://dx.doi.org/10.4236/pp.2014.51017
- Buckley, M.M., Goa, K.L., Price, A.H. and Brogden, R.N. (1989) Probucol. A Reappraisal of Its Pharmacological Properties and Therapeutic Use in Hypercholesterolaemia. Drugs, 37, 761-800. http://dx.doi.org/10.2165/00003495-198937060-00002
- Jeon, H., Lee, S., Kim, T.-E., Yoon, S.H., Shin, S.-G., Jang, I.-J. and Yu, K.-S. (2011) Pharmacokinetics and Tolerability of Probucol after Multiple Oral Administrations in Healthy Volunteers. International Journal of Clinical Pharmacology and Therapeutics, 49, 688-695. http://dx.doi.org/10.5414/CP201525
- Klein, L. (1981) QT-Interval Prolongation Produced by Probucol. Archives of Internal Medicine, 141, 1102-1103. http://dx.doi.org/10.1001/archinte.1981.00340080138037
- Matsuhashi, H., Onodera, S., Kawamura, Y., Hasebe, N., Kohmura, C., Yamashita, H. and Tobise, K. (1989) Probucol-Induced QT Prolongation and Torsades de Pointes. Japanese Journal of Medicine, 28, 612-615. http://dx.doi.org/10.2169/internalmedicine1962.28.612
- Gohn, D.C. and Simmons, T.W. (1992) Polymorphic Ventricular Tachycardia (Torsade de Pointes) Associated with the Use of Probucol. The New England Journal of Medicine, 326, 1435-1436. http://dx.doi.org/10.1056/NEJM199205213262117
- Tamura, M., Ueki, Y., Ohtsuka, E., Oribe, M., Seita, M., Oribe, K. and Ito, M. (1994) Probucol-Induced QT Prolongation and Syncope. Japanese Circulation Journal, 58, 374-377. http://dx.doi.org/10.1253/jcj.58.374
- Guo, J., Massaeli, H., Li, W., Xu, J., Luo, T., Shaw, J., Kirshenbaum, L.A. and Zhang, S. (2007) Identification of IKr and Its Trafficking Disruption Induced by Probucol in Cultured Neonatal Rat Cardiomyocytes. The Journal of Pharmacology and Experimental Therapeutics, 321, 911-920. http://dx.doi.org/10.1124/jpet.107.120931
- Guo, J., Li, X., Shallow, H., Xu, J., Yang, T., Massaeli, H., Li, W., Sun, T., Pierce, G.N. and Zhang, S. (2011) Involvement of Caveolin in Probucol-Induced Reduction in hERG Plasma-Membrane Expression. Molecular Pharmacology, 79, 806-813. http://dx.doi.org/10.1124/mol.110.069419
- Guo, J., Wang, T., Yang, T., Xu, J., Li, W., Fridman, M.D., Fisher, J.T. and Zhang, S. (2011) Interaction between the Cardiac Rapidly (IKr) and Slowly (IKs) Activating Delayed Rectifier Potassium Channels Revealed by Low K+-Induced hERG Endocytic Degradation. The Journal of Biological Chemistry, 286, 34664-34674. http://dx.doi.org/10.1074/jbc.M111.253351
Abbreviations
APD90, Action Potential Duration at 90% Repolarization b.i.d., bis in die (twice a day)
CHO, Chinese Hamster Ovary Cmax, Maximum Concentration DMEM, Dulbecco’s Modified Eagle’s Medium DMSO, Dimethyl Sulfoxide FBS, Fetal Bovine Serum FPD, Field Potential Duration FPDc, Corrected FPD HEK, Human Embryonic Kidney hERG, Human ether-à-go-go-Related Gene hES-CMC, Human Embryonic Stem Cell-Derived Cardiomyocyte Cluster ISI, Inter-Spike Interval i.v., Intravenous NEAA, Non-Essential Amino Acids q.d., quaque die (every day)
QTc, Corrected QT TdP, Torsades de Pointes