Pharmacology & Pharmacy
Vol.3 No.2(2012), Article ID:18713,8 pages DOI:10.4236/pp.2012.32023
Hypersensitive Inhibition of the Proliferation of Cells with Mutated DNA Repair-Related Genes by the Catalytic Topoisomerase II Inhibitor 20-O-IngenolEZ
![]()
1Department of Chemistry, College of Humanities and Sciences, Nihon University, Tokyo, Japan; 2School of Pharmacy, Nihon University, Chiba, Japan.
Email: *miyata@chs.nihon-u.ac.jp
Received January 9th, 2012; revised February 25th, 2012; accepted March 6th, 2012
Keywords: Catalytic Topo II Inhibitor; DNA Damage Checkpoint; DNA Repair-Related Genes; Ingenol Compound
ABSTRACT
We previously reported that many ingenol compounds derived from Euphoria kansui exhibit topoisomerase inhibitory activity. 20-O-ingenolEZ in these compounds exerted inhibitory effects on both topoisomerase II (topo II) activity and cell proliferative activity. Topoisomerase II inhibitors can be divided into the poison and catalytic inhibitor types and 20-O-ingenolEZ is a catalytic inhibitor and inhibits topo IIα through inhibition of ATPase activity, but induces topo II-mediated DNA damage and apoptosis in BLM−/− DT40 cells through the induction of the DNA damage checkpoint, similar to the poison type inhibitor adriamycin. The ATPase inhibitor of topo II ICRF-193 also showed poison-like characteristics in the same cell line. However, the inhibitory effects of ICRF-193 on the proliferation of BLM−/− DT40 cells differed from those of 20-O-ingenolEZ, as did the specificity of its inhibition of the proliferation of other cell lines. 20-O-ingenolEZ showed hypersensitive inhibition of the proliferation of MCF-7 cells and BLM−/− DT40 cells with mutated DNA repair-related genes.
1. Introduction
Topoisomerase II (topo II) is essential for the survival of proliferating cells, and it is involved in recombination, chromosome condensation, and decatenation of sister chromatids before anaphase of mitosis [1]. In cancer chemotherapy, topo II is a major target for various anticancer drugs. These drugs can be divided into two types based on their mode of action. One type is the topo II poisons, so named because of their severe cytotoxicity resulting from their ability to stabilize enzyme-DNA covalent complexes and activates DNA damage checkpoint [2,3]. Topo II catalytic inhibitors do not stabilize topo II-DNA covalent complexes. Instead, these compounds block the enzyme before DNA cleavage or at the last step of the catalytic cycle after religation [4,5]. An example of this latter type of inhibitors is ICRF-193 (bisdioxopiperazine), which blocks the opening of an already closed clamp by inhibiting ATPase activity of the enzyme and preventing DNA decatenation of the replicated chromosomes by it, thus arresting cell proliferation [6,7]. These catalytic inhibitors inhibit topo II before the transient double-strand breakage step and activate the decatenation checkpoint [8,9]. The topo II poisons have been developed as anticancer agents for clinical use [10]. However, they function by stabilizing the state in which the enzyme introduced DSBs [11,12]. Despite their broad anticancer activity, the use of topo II poisons is limited by their narrow therapeutic window, since the damage to healthy cells that divided more rapidly, such as bone marrow cells and the gut lining and tissues is almost inevitable. Thus, new types of potent inhibitors of topo II that induce the DNA decatenation checkpoint are desired [13, 14].
We previously reported that 20-O-ingenolEZ functions as a catalytic inhibitor and inhibits the ATPase activity of topo IIα and induces G2 arrest of MMT cells [15]. However, treatment of BLM−/− DT40 cells with 20-O-ingenolEZ induces DNA damage signaling and apoptosis [16]. Although the inhibition of cell proliferation through the induction of topo II decatenation checkpoint is weak than that by topo II damage checkpoint [17-19], catalytic inhibitors have low DNA truncation toxicity [20,21] and an inefficient decatenation checkpoint for stem and progenitor cells [22]. Furthermore, the catalytic inhibitor ICRF-193 was showed to be hypersensitive to inhibition of cell proliferation of cancer cells with impaired decatenation checkpoint function and to induce apoptosis of these specific cells [23,24]. Topo II catalytic inhibitors which induces DNA damage signaling and apoptosis is useful as new types of potent inhibitors of topo II. More studies are required to identify particular cell lines responsive to 20-O-ingenolEZ and ICRF-193. In this study, an assay for detecting topo II poisons or catalytic inhibitors was preformed using a compound structurally distinct but related to 20-O-ingenolEZ. Furthermore, since the specificity of inhibition of the proliferation of various cell lines by similar compounds may not be correlated, the specificity of cell proliferation inhibition by 20-OingenolEZ and ICRF-193 was compared using other mutated cell lines, although the mechanism of inhibition of topo II activities was the same for the two compounds. The selection of decatenation or DNA damage signaling by ICRF-193 and 20-O-ingrnolEZ depended on the cell type, and ICRF-193 showed the same poison-like effects on BLM−/− DT40 cells. However, inhibition of cell prolixferation differed between the two catalytic inhibitors in several cell lines. 20-O-ingenolEZ displayed hypersensitive inhibition of proliferation of cells with mutated DNA repair-related genes and induced DNA damage signaling and apoptosis.
2. Materials and Methods
2.1. Cell Lines and Cell Proliferation
BLM−/− DT40 cells were provided by RIKEN BRC through the National Bio-Resource Project of the MEXT, Japan. BLM−/− DT40 cells were grown in RPMI 1640 medium containing 50 μM of mercaptoethanol, 10% fetal bovine serum (FBS), and 1% chicken serum. Rat testicular tumor (LC540) cells and mouse normal 3T3-Swiss albino (3T3) cells were grown in Minimum Essential Medium (MEM) supplemented with 10% FBS. BT-474 cells were grown in Hybri-Care medium containing 10% FBS. MCF-7 cells were grown in MEM supplemented with 25 μM insulin, 1 μM sodium pyruvate, 10% nonessential amino acids, and 10% FBS. The cells were then transplanted on microculture plates. The diterpene compound 20-O-(2¢(E),4¢(Z)-decadienoyl) ingenol (20-O-ingenolEZ) was dissolved in dimethyl sulfoxide. After appropriate dilution, the compound was added to 100 μl of medium and the cells were incubated for 24 h at 37˚C in 95% air and 5% CO2. Cell growth in the presence or absence of the compound was determined using the 3- (4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide (MTT) assay [25] (Roche Applied Science). The optical density of each well was measured at 620 nm using a plate reader (Amersham).
2.2. Topo II-Mediated Supercoiled pBR322 Relaxation
The activity of the compounds on relaxation induced by DNA topo IIα (human recombinant in E. coli, Topo GEN, Inc. USA) was determined by measuring the conversion of supercoiled pBR322 plasmid DNA (TOYOBO, Tokyo) to its relaxed form. The reaction mixture contained 50 mM Tris-HCl (pH 8.0), 150 mM NaCl, 10 mM MgCl2, 500 μM DTT, 30 μg/ml BSA, 5 mM ATP, 90 ng pBR322, 1 U of enzyme and different concentrations of the compounds in a total volume of 20 μl. After incubation for 9.5 min at 37˚C, the mixture was subjected to electrophoresis on a 1% agarose gel. After electrophoresis, the gel was stained with ethidium bromide and visualized under UV light.
2.3. ATPase Assay
Hydrolysis catalyzed by human topo IIα was examined by measuring the liberating of phosphate, as described by Cogan et al. [26]. The reaction mixture contained 50 mM Tris-HCl (pH 8.0), 150 mM NaCl, 5 mM ATP, 500 μM DTT, 30 μg/ml BSA, 180 ng pBR322 and 1 U human topo IIα. After 9.5 min, 200 μl of reagent containing 0.23 mg/ml malachite green, 10.6 mg/ml ammonium molybdate, 6% H2SO4, and 0.02% Tween 20 was added, and the mixture was incubated for 5 min at room temperature for color development. Absorbance was measured at 620 nm using a microplate reader.
2.4. Western Blotting
BLM−/− DT40 cells were cultured for 24 h in the presence of 200 μM 20-O-ingenolEZ, 5 μM ICRF-193, or 0.9 μM adriamycin, washed in phosphate-buffered saline (PBS), suspended in a lysis buffer [20 mM HEPES (pH 7.8), 10 mM NaCl, 1.5 mM MgCl2, 0.2 mM EDTA, 0.1% TritonX100, 20% glycerol, and 1 mM DTT supplemented with protease inhibitors] and lysed. After centrifugation at 2,100 rpm for 4 min, the precipitate was resuspended in lysis buffer containing 500 mM NaCl instead of 10 mM NaCl and placed on ice for 30 min. The suspension was centrifuged at 15,000 rpm for 15 min at 4˚C. Twenty μg of nuclear protein was subjected to electrophoresis on 15% SDS-polyacrylamide gels, and transferred to a polyvinylidene difluoride membrane. Blots were prepared using anti-γ-H2AX antibody (Millipore, Billerica, MA, USA) followed by detection with an enhanced chemiluminescence system.
2.5. Apoptotic Assay
BLM−/− DT40 cells were cultured on coverslips for 24 h in the presence of 200 μM 20-O-ingenolEZ, 5 μM ICRF- 193, or 0.9 μM adriamycin. Cells were washed three times with PBS for 5 min. Apoptotic cells and nuclei were stained for 10 min at room temperature with 4,6- diamino-2-phenyl indole (DAPI, at 1 μg/ml PBS) and detected by fluorescence microscopy.
3. Results
3.1. DNA Relaxation Assay of 20-O-IngenolEZ-Treated Supercoiled pBR322 Plasmid DNA
Effects of 20-O-ingenolEZ and ICRF-193 on the strand passage activity of topo II were determined by their effects on enzyme-mediated supercoiled pBR322 relaxation. As shown in Figure 1, 20 μM 20-O-ingenolEZ almost completely inhibited this reaction and 40 μM 20-OingenolEZ completely inhibited it (Figure 1(a)), while 10 μM ICRF-193 completely inhibited this reaction (Figure 1(b)). ICRF-193 was more effective against human topo II than 20-O-ingenolEZ.
3.2. ATPase Inhibition Assay of Topo II with 20-O-IngenolEZ
To further investigate the inhibition of eukaryotic topo II by 20-O-ingenolEZ, we studied the effects of 20-O-ingenolEZ and ICRF-193 on DNA-stimulated ATPase activy of human topo II. 20-O-ingenolEZ or ICRF-193 inhibited ATPase domains of human topo II (Figure 2) and inhibited ATPase activity by a maximum of approximately 80% of that of the control. There were 50% reduction in ATP hydrolysis with about 50 μM 20-O-ingenolEZ and 20 μM ICRF-193. Although, the function of 20- O-ingenolEZ appeared to be similar to ICRF-193, ICRF- 193 had higher inhibitory activity than 20-O-ingenolEZ.
3.3. Effects on the Proliferation of Cancer Cells
To investigate the inhibitory effects of 20-O-ingenolEZ and ICRF-193 on the proliferation of cancer cell lines, a concentration-response range (0.5 and 200 μM) was established using an exposure time of 24 h. Since, we already knew that 20-O-ingenolEZ exhibited a hypersensitive inhibitory effect on the proliferation of BLM−/− DT40 cells with mutated DNA repair genes compared with other cell lines, we first compared the effects of 20-OingenolEZ and ICRF-193 on the proliferation of BLM−/− DT40 cells [11]. The proliferation of BLM−/− DT40 cells was not inhibited by 20-O-ingenolEZ at concentrations of 2 - 60 μM, but was completely and immediately inhibited at concentrations higher than 100 μM (Figure 3(a)). The effects of ICRF-193 on BLM−/− DT40 were different. The proliferation of BLM−/− DT40 cells was gradually inhibited as the concentration of ICRF-193 increased,
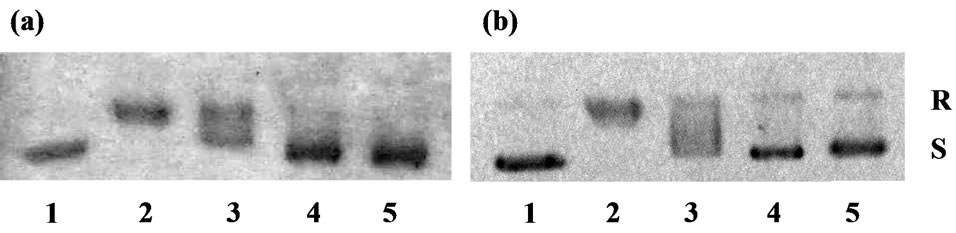
Figure 1. Effects of 20-O-ingenolEZ and ICRF-193 on human topo II activity. To determine inhibition of relaxation of the supercoils, pBR322 DNA was incubated with topo IIα. Each reaction mixture (20 μl) contained 90 ng pBR322 plasmid DNA, 1 U human topo IIα and 20, 40, 60 μM 20-OingenolEZ or 5, 10, 15 μM ICRF-193. After incubation for 9.5 min at 37˚C, the mixture was subjected to electrophoresis on an agarose gel. (a) Lanes 1, no enzyme; Lane 2, with enzyme; Lanes 3-5, with 20, 40, 60 μM 20-O-ingenolEZ; (b) Lanes 1, no enzyme; Lanes 2, with enzyme; Lanes 3-5, with 5, 10, 15 μM ICRF-193. Abbreviations: R, Relaxed DNA; S, supercoiled DNA.
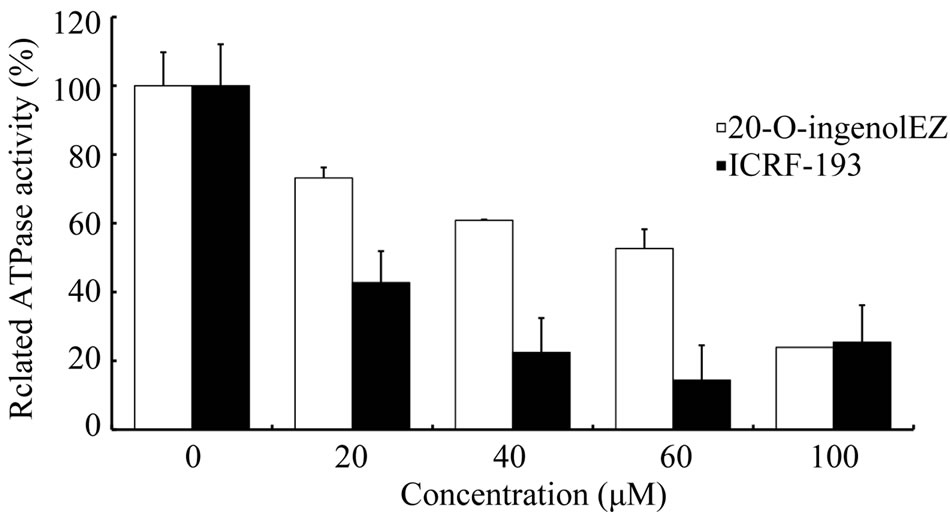
Figure 2. Effects of 20-O-ingenolEZ and ICRF-193 on the ATPase activity of topo II. DNA-dependent ATPase activity was measured in a reaction mixture containing 50 mM Tris-HCl (pH 8.0), 150 mM NaCl, 5 mM ATP, 500 μM DTT, 30 μg/ml BSA, 180 ng pBR322, and 1 U human topo IIα at various 20-O-ingenolEZ and ICRF-193 concentrations. Free phosphate was measured with a malachite green reagent. Relative ATPase activity was normalized against activity under conditions without 20-O-ingenolEZ and ICRF-193.
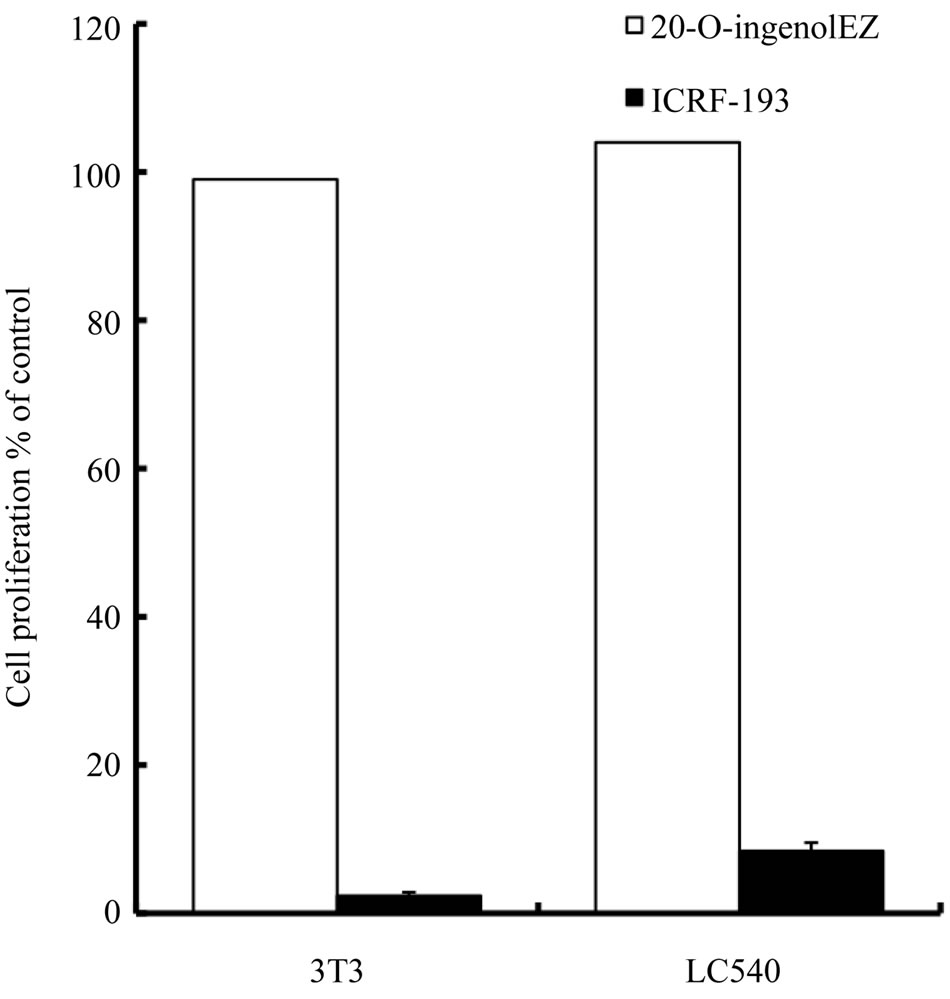
reaching a maximum inhibition of about 80% - 90% during 50 - 200 μM (Figure 3(b)). We next compared the effects of high concentration of 20-O-ingenolEZ and ICRF-193 on rodent cell lines containing normal mouse embryo 3T3 cells. Although the proliferation of 3T3 and LC540 cell lines was not inhibited by 20-O-ingenolEZ over a concentration range of 2 - 200 μM, both cell types were completely inhibited by 200 μM ICRF-193 (Figure 4(a)). Finally, we compared the effects of 20-O-ingenolEZ and ICRF-193 on a human breast cancer cell line containing BT-474 and MCF-7 cells, which are mutated in HER2 or RAD51C, respectively. Although 200 μM 20-O-ingenolEZ completely inhibited the proliferation of MCF-7 cells, it did not inhibit the proliferation of BT- 474 cells (Figure 4(b)). An ICRF-193 concentration of 200 μM weakly inhibited both human breast cancer cell lines (Figure 4(b)).
 (a)
(a) (b)
(b)
Figure 3. Effects of 20-O-ingenolEZ on the proliferation of BLM−/− DT40 cells. BLM−/− DT40 cells were cultured in microplates at 37˚C for 24 h in the presence or absence of 2, 20, 60, 100 and 200 μM 20-O-ingenolEZ or 0.5, 5, 50, 100 and 200 μM ICRF-193. Relative cell growth was determined by MTT assay. The cell growth in untreated cells was set as 100% and that of BLM−/− DT40 cells treated with 2, 20, 60, 100 and 200 μM 20-O-ingenolEZ or 0.5, 5, 50, 100 and 200 μM ICRF-193 was expressed relative to the level in untreated BLM−/− DT40 cells (100%). Expressions were assessed in triplicate and data are shown as means ± SD. (a) 20-O-ingenolEZ and (b) ICRF-193.
3.4. Effects of 20-O-IngenolEZ on the Phosphorylation of H2AX
We also observed the appearance of phosphorylated H2AX (γ-H2AX), which serves as a marker of DNA double strand breaks (DSBs) [27], using BLM−/− DT40 cells. Adriamycin is known to stabilize topo II DNAcovalent complexes, and DNA damage is induced through numerous signaling pathways. γ-H2AX was visualized as a band stained with anti-γ-H2AX in the nuclei of adriamycin-treated BLM−/− DT40 cells (Figure 5, lane 3). We observed a similar induction of DSBs in 20-O-ingenolEZand ICRF-193-treated BLM−/− DT40 cells (Figure 5, lanes 2 and 4).
3.5. Effects of 20-O-IngenolEZ and ICRF-193 on Apoptosis
Since both 20-O-ingenolEZand ICRF-193-treated BLM−/− DT40 cells exhibited DSBs, induction of apoptosis in BLM−/− DT40 cells was observed using both catalytic inhibitors. The morphological characteristics of the apoptotic cells in each sample were determined based on staining with 4,6-diamino-2-phenyl indole (DAPI). After
 (a)
(a) (b)
(b)
Figure 4. Effects of 20-O-ingenolEZ on the proliferation of various cell lines. 3T3, LC540, MCF-7 and BT-474 cells were cultured in microplates at 37˚C for 24 h in the presence or absence of 200 μM 20-O-ingenolEZ or ICRF-193. Relative cell growth was determined by MTT assay. The cell growth in untreated cells was set as 100% and that of various cells treated with 200 μM 20-O-ingenolEZ or ICRF- 193 was expressed relative to the level in the untreated cells (100%). Expressions were assessed in triplicate and data are shown as means ± SD. (a) 3T3 cell, LC540 cell and (b) MCF-7 cell, BT-474 cell.

Figure 5. Effects of 20-O-ingenolEZ on phosphorylation of H2AX in BLM−/− DT40 cells. For immunoblotting of γ- H2AX in BLM−/− DT40 cells, cells were cultured in the present of 200 μM 20-O-ingenolEZ, 5 μM ICRF-193, or 0.9 μM adriamycin for 24 h. The nuclear protein fraction (20 μg) was resolved by SDS-PAGE, followed by western blotting and chemiluminescence detection. γ-H2AX was detected using a specific antibody against γ-H2AX. Lane 1, control; Lane 2, 20-O-ingenolEZ; Lane 3, adriamycin; Lane 4, ICRF- 193.
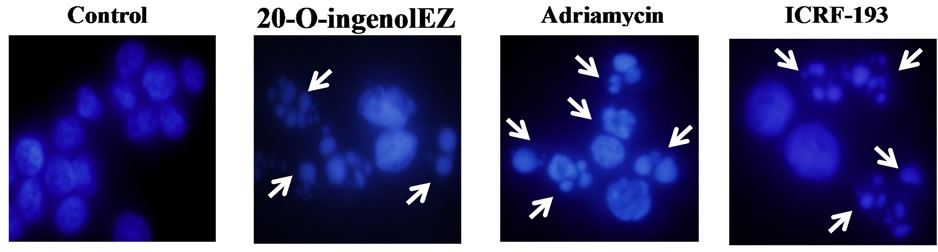
24 h of treatment with 0.9 μM adriamycin, BLM−/− DT40 cells were stained with DAPI (Figure 6(a)), and apoptosis was found to be induced in approximately 60% of cells (Figure 6(b)). 20-O-ingenolEZ-treated BLM−/− DT40 cells also exhibited morphological characteristics of apoptosis similar to those observed in adriamycin-treated BLM−/− DT40 cells (Figure 6). ICRF-193-treated BLM−/− DT40 cells were also apoptotic (Figure 6).
4. Discussion
We compared the effects of 20-O-ingenolEZ and ICRF- 193 on topo II. Both compounds inhibited ATPase activity by a maximum of approximately 80% of that of the controls. However, 20-O-ingenolEZ was less potent than ICRF-193. ICRF-193 inhibited the proliferation of BLM−/− DT40 cell and induced DNA damage signaling and apoptosis in a manner similar to that of 20-O-ingenolEZ, but it had different specificity with respect to the inhibition of proliferation of various cell lines. 20-O-ingenolEZ showed hypersensitivity to cells with mutated DNA repair-related genes, such as BLM−/− DT40 cells containing the gene mutated in Bloom’s syndrome and MCF-7 cells containing the RAD51C-ATXN7 fusion gene. This indicates that 20-O-ingenonEZ and ICRF-193 may differ in their function.
ICRF-193 treatment delayed the G2/M transition of the cell cycle [28,29] and the nature of this G2 delay has been proposed as a “decatenation checkpoint” where cells monitor chromatide catenation status after DNA replication and inhibit progression to mitosis until decatenation of the chromatids by topo II is accurate [30]. Many studies have suggested that the decatenation checkpoint is distinct from the DNA damage checkpoint. Although, phosphorylation of H2AX was not induced when MMT cells were treated with 50 μM ICRF-193 or 200 μM 20-O-ingenolEZ for 24 h, at a concentration that completely inhibited cell proliferation, it was induced when MMT cells were treated with 0.9 μM adriamycin
 (a)
(a)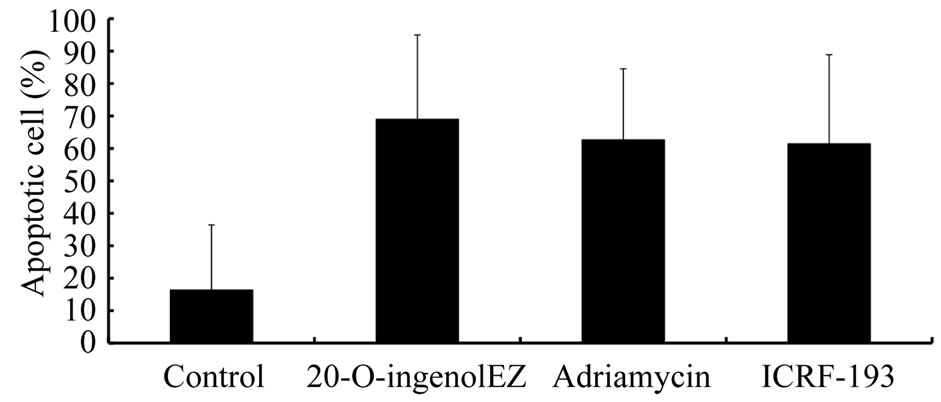 (b)
(b)
Figure 6. Effects of 20-O-ingenolEZ on apoptosis in BLM−/− DT40 cells. BLM−/− DT40 cells were treated at 37℃ with 200 μM 20-O-ingenolEZ, 5 μM ICRF-193, 0.9 μM adriamycin, or control for 24 h. Apoptosis was detected by DAPI staining and arrows in the image indicate apoptotic cells after 24 h treatment with 20-O-ingenolEZ, adriamycin, or ICRF-193 (a). The percentage of apoptotic BLM−/− DT40 cells after 20-O-ingenolEZ, ICRF-193, and adriamycin treatments is shown (b).
[16,31]. However, we have demonstrated that 20-O-ingenolEZ is a novel topo II catalytic inhibitor that induces DNA damage signaling in BLM−/− DT40 cells. This inhibition induces the phosphorylation of H2AX in response to agents that introduce topo II-mediated DNA damage signaling. ICRF-193 has been extensively analyzed as a topo II catalytic inhibitor in studies of the functions of topo II [2-5,28-30], and several recent observations have suggested that ICRF-193 may induce DNA damage [20,32,33]. Therefore, we studied damage signaling induced by ICRF-193, which inhibited the proliferation of the BLM−/− DT40 cells in this studies. We also observed that the two types of topo II catalytic inhibitors activated DNA damage signaling and apoptosis in BLM−/− DT40 cells. With regard to the mechanism of induction of damage signaling and apoptosis, topo II poison such as VP- 16 (etoposide) has been shown to induce preferential degradation of topo IIβ through a proteasome pathway, which is responsible for the exposure of topo IIβ-mediated DSBs [34]. Furthermore, it has been revealed that ICRF-193 can also arrest transcription and triggers proteosomal degradation of topo IIβ. It is believed that in the presence of ICRF-193 and ATP, topo IIβ is trapped as a closed circular clamp on DNA and the topo II circular clamp triggers 26S proteosome-dependent degradation of topo IIβ [35]. DSBs induced by proteasome degradation can be repaired by homologous recombination and /or by nonhomologous endojoining [17]. If DNA checkpoint and repair responses cannot restore genetic stability, then cells may undergo apoptosis [36]. Since BLM−/− DT40 cells with mutated DNA repair gene cannot repair chromosome damaged as a result of failed decatenation, these inhibitors may induce apoptosis in BLM−/− DT40 cells by proteasomal degradation.
If 20-O-ingenolEZ induces DNA repair signaling through decatenation checkpoint in BIM−/− DT40 cells, it may be hypersensitive to cells with mutated DNA repair-related genes and induce apoptosis [23,24,36]. Although 20-O-ingenolEZ inhibited cell proliferation on BLM−/− DT40 cells, it did not inhibit the proliferation of WRN−/− DT40 cells that have impaired RecQ helicase [16]. Therefore, we studied the effects of 20-O-ingenolEZ and ICRF-193 on proliferation on cell lines with mutated DNA repair-related genes that function together with BLM. BLM forms a complex with the recombination protein RAD51C. The interaction between BLM and RAD51C provides the first direct molecular link in the pathway for recombinational repair in human cells [37- 39]. In the MCF-7 breast cancer cell line, RAD51CATXN7 fusion gene breakpoints have been confirmed [40,41]. 20-O-ingenolEZ showed hypersensitivity to inhibition of cell proliferation of MCF-7 cells compared with that of BT-474 breast cancer cell, which overexpressed ErbB mRNA [42]. In a recent study, RAD51C was confirmed as a breast and ovarian cancer susceptibility gene [43]. We revealed that a catalytic inhibitor inducing a decatenation checkpoint, 20-O-ingenolEZ, inhibited the proliferation of BLM−/− DT40 [16] and MCF-7 cells with mutated DNA repair-related gene; it was hypersensitive to these cells compared to BT-474 breast cancer cells. Thus, compounds that target the induction of the decatenation checkpoint may selectively target repair-deficient cancer cells such as RAD51C. Further drug discovery efforts relating to ingenol compounds are warranted and such drugs could potentially be developed as anticancer therapies.
5. Acknowledgements
This investigation was supported by the “Academic Frontier (2007-2009)” Projects for private universities, with a matching fund subsidy from MEXT (Ministry of Education, Culture, Sports, Science and Technology of Japan).
REFERENCES
- J. C. Wang, “Cellular Roles of DNA Topoisomerases: A Molecular Perspective,” Nature Reviews Molecular Cell Biology, Vol. 3, No. 6, 2002, pp. 430-440. doi:10.1038/nrm831
- S. J. Froelich-Ammon and N. Osheroff, “Topoisomerase Poisons: Harnessing the Dark Side of Enzyme Mechanism,” Journal of Biological Chemistry, Vol. 270, No. 37, 1995, pp. 21429-21432. doi:10.1074/jbc.270.37.21429
- D. A. Burden and N. Osheroff, “Mechanism of Action of Eukaryotic Topoisomerase II and Drugs Targeted to the Enzyme,” Biochimica et Biophysica Acta, Vol. 1400, No. 1-3, 1998, pp. 139-154.
- T. Andoh and R. Ishida, “Catalytic Inhibitors of DNA Topoisomerase II,” Biochimica et Biophysica Acta, Vol. 1400, No. 1-3, 1998, pp. 155-171.
- A. K. Larsen, A. E. Escargueil and A. Skladanowski, “Catalytic Topoisomerase II Inhibitors in Cancer Therapy,” Pharmacology & Therapeutics, Vol. 99, No. 2, 2003, pp. 167-181. doi:10.1016/S0163-7258(03)00058-5
- J. Roca, R. Ishida and J. M. Berger, “Antitumor Bisdioxopiperazines Inhibit Yeast DNA Topoisomerase II by Trapping the Enzyme in the Form of a Closed Protein Clamp,” Proceedings of the National Academy of Sciences of the United States of America, Vol. 91, No. 5, 1994, pp.1781-1785. doi:10.1073/pnas.91.5.1781
- T. Hu, H. Sage and T. S. Hsieh, “ATPase Domain of Eukaryotic DNA Topoisomerase II. Inhibition of ATPase Activity by the Anti-Cancer Drug Bisdioxopiperazine and ATP/ADP-Induced Dimerization,” Journal of Biological Chemistry, Vol. 277, No. 8, 2002, pp. 5944-5951. doi:10.1074/jbc.M111394200
- M. S. Hossain, N. Akimitsu, T. Takaki, H. Hirai and K. Sekimizu, “ICRF-193, a Catalytic Inhibitor of DNA Topoisomerase II, Inhibits Re-Entry into the Cell Division Cycle from Quiescent State in Mammalian Cells,” Genes Cells, Vol. 7, No. 3, 2002, pp. 285-294. doi:10.1046/j.1365-2443.2002.00521.x
- M. Damelin and T. H. Bestor, “The Decatenation Checkpoint,” British Journal of Cancer, Vol. 96, No. 2, 2007, pp. 201-205. doi:10.1038/sj.bjc.6603537
- K. R. Hande, “Clinical Applications of Anticancer Drugs Targeted to Topoisomerase II,” Biochimica et Biophysica Acta, Vol. 1400, No. 1-3, 1998, pp. 173-184.
- W. E. Ross, D. Glaubiger and K. W. Kohn, “Qualitative and Quantitative Aspects of Intercalator-Induced DNA Strand Breaks,” Biochimica et Biophysica Acta, Vol. 562, No. 1, 1979, pp. 41-50.
- K. M. Tewey, T. C. Rowe, L. Yang, B. D. Halligan and L. F. Liu, “Adriamycin-Induced DNA Damage Mediated by Mammalian DNA Topoisomerase II,” Science, Vol. 226, No. 4673, 1984, pp. 466-468. doi:10.1126/science.6093249
- S. J. Haggarty, K. M. Koeller, T. R. Kau, P. A. Silver, M. Roberge and S. L. Schreiber, “Small Molecule Modulation of the Human Chromatid Decatenation Checkpoint,” Chemistry & Biology, Vol. 10, No. 12, 2003, pp. 1267- 1279. doi:10.1016/j.chembiol.2003.11.014
- P. Chène, J. Rudloff, J. Schoepfer, P. Furet, P. Meier, Z. Qian, J. M. Schlaeppi, R. Schmitz and T. Radimerski, “Catalytic Inhibition of Topoisomerase II by a Novel Rationally Designed ATP-Competitive Purine Analogue,” BMC Chemical Biology, Vol. 9, No. 1, 2009, pp. 1-16. doi:10.1186/1472-6769-9-1
- C. Yoshida, K. Hishiyama, K. Miyazaki, M. Watanabe, M. Kanbe, Y. Yamada, K. Matsuzaki, K. Miyashita, S. Kitanaka and S. Miyata, “Analysis of Inhibition of Topoisomerase IIalpha and Cancer Cell Proliferation by IngenolEZ,” Cancer Science, Vol. 101, No. 2, 2010, pp. 374-378. doi:10.1111/j.1349-7006.2009.01408.x
- M. Watanabe, Y. Kamada, K. Miyazaki, S. Mizoguchi, K. Matsuzaki, S. Kitanaka and S. Miyata, “20-O-IngenolEZ, a Catalytic Topoisomerase II Inhibitor, Specifically Inhibits Cell Proliferation and Induces Double-Strand DNA Breaks in BLM−/− Cells,” Medicinal Chemistry Communications, Vol. 2, No. 9, 2011, pp. 824-827. doi:10.1039/c0md00252f
- N. Adachi, H. Suzuki, S. Iiizumi and H. Koyama, “Hypersensitivity of Nonhomologous DNA End-Joining Mutants to VP-16 and ICRF-193: Implications for the Repair of Topoisomerase II-Mediated DNA Damage,” Journal of Biological Chemistry, Vol. 278, No. 38, 2003, pp. 35897- 35902. doi:10.1074/jbc.M306500200
- C. Pérez, N. E. Vilaboa, L. García-Bermejo, E. de Blas, A. M. Creighton and P. Aller, “Differentiation of U-937 Promonocytic Cells by Etoposide and ICRF-193, Two Antitumour DNA Topoisomerase II Inhibitors with Different Mechanisms of Action,” Journal of Cell Science, Vol. 110, No. 3, 1997, pp. 337-343.
- T. Marple, T. M. Kim and P. Hasty, “Embryonic Stem Cells Deficient for Brca2 or Blm Exhibit Divergent Genotoxic Profiles That Support Opposing Activities during Homologous Recombination,” Mutation Research, Vol. 602, No. 1-2, 2006, pp. 110-120. doi:10.1016/j.mrfmmm.2006.08.005
- N. Pastor, I. Domínguez, S. Mateos and F. Cortés, “A Comparative Study of Genotoxic Effects of Anti-Topoisomerase II Drugs ICRF-193 and Bufalin in Chinese Hamster Ovary Cells,” Mutation Research, Vol. 515, No. 1-2, 2002, pp. 171-180.
- L. H. Jensen, K. C. Nitiss, A. Rose, J. Dong, J. Zhou, T. Hu, N. Osheroff, P. B. Jensen, M. Sehested and J. L. Nitiss, “A Novel Mechanism of Cell Killing by Anti-Topoisomerase II Bisdioxopiperazines,” Journal of Biological Chemistry, Vol. 275, No. 3, 2000, pp. 2137-2146. doi:10.1074/jbc.275.3.2137
- M. Damelin, Y. E. Sun, V. B. Sodja and T. H. Bestor, “Decatenation Checkpoint Deficiency in Stem and Progenitor Cells,” Cancer Cell, Vol. 8, No. 6, 2005, pp. 479- 484. doi:10.1016/j.ccr.2005.11.004
- A. Franchitto, J. Oshima and P. Pichierri, “The G2-Phase Decatenation Checkpoint Is Defective in Werner Syndrome Cells,” Cancer Research, Vol. 63, No. 12, 2003, pp. 3289-3295.
- T. Nakagawa, Y. Hayashita, K. Maeno, A. Masuda, N. Sugito, H. Osada, K. Yanagisawa, H. Ebi, K. Shimokata and T. Takahashi, “Identification of Decatenation G2 Checkpoint Impairment Independently of DNA Damage G2 Checkpoint in Human Lung Cancer Cell Lines,” Cancer Research, Vol. 64, No. 14, 2004, pp. 4826-4832. doi:10.1158/0008-5472.CAN-04-0871
- M.C. Alley, D.A. Scudiero, A. Monks, M. L. Hursey, M. J. Czerwinski, D. L. Fine, B. J. Abbott, J. G. Mayo, R. H. Shoemaker and M. R. Boyd, “Feasibility of Drug Screening with Panels of Human Tumor Cell Lines Using a Microculture Tetrazolium Assay,” Cancer Research, Vol. 48, No. 3, 1988, pp. 589-601.
- E. B. Cogan, G. B. Birrell and O. H. Griffith, “A Robotics-Based Automated Assay for Inorganic and Organic Phosphates,” Analytical Biochemistry, Vol. 271, No. 1, 1999, pp. 29-35. doi:10.1006/abio.1999.4100
- E. P. Rogakou, D. R. Pilch and A. H. Orr, “DNA Double-Stranded Breaks Induce Histone H2AX Phosphorylation on Serine 139,” Journal of Biological Chemistry, Vol. 273, No. 10, 1998, pp. 5858-5868. doi:10.1074/jbc.273.10.5858
- P. R. Andreassen, F. B. Lacroix and R. L. Margolis, “Chromosomes with Two Intact Axial Cores Are Induced by G2 Checkpoint Override: Evidence That DNA Decatenation Is Not Required to Template the Chromosome Structure,” Journal of Cell Biology, Vol. 136, No. 1, 1997, pp. 29-43. doi:10.1083/jcb.136.1.29
- C. S. Downes, D. J. Clarke, A. M. Mullinger, J. F. Giménez-Abián, A. M. Creighton and R. T. Johnson, “A Topoisomerase II-Dependent G2 Cycle Checkpoint in Mammalian Cells,” Nature, Vol. 372, No. 6505, 1994, pp. 467-470. doi:10.1038/372467a0
- P. B. Deming, C. A. Cistulli, H. Zhao, P. R. Graves, H. Piwnica-Worms, R. S. Paules, C. S. Downes and W. K. Kaufmann, “The Human Decatenation Checkpoint,” Proceedings of the National Academy of Sciences of the United States of America, Vol. 98, No. 21, 2001, pp. 12044-12049. doi:10.1073/pnas.221430898
- K. Luo, J. Yuan, J. Chen and Z. Lou, “Topoisomerase II Alpha Controls the Decatenation Checkpoint,” Nature Cell Biology, Vol. 11, No. 2, 2009, pp. 204-210. doi:10.1038/ncb1828
- I. Park and H. K. Avraham, “Cell Cycle-Dependent DNA Damage Signaling Induced by ICRF-193 Involves ATM, ATR, CHK2, and BRCA1,” Experimental Cell Research, Vol. 312, No. 11, 2006, pp. 1996-2008. doi:10.1016/j.yexcr.2006.02.029
- H. M. Robinson, S. Bratlie-Thoresen, R. Brown and D. A. Gillespie, “Chk1 Is Required for G2/M Checkpoint Response Induced by the Catalytic Topoisomerase II Inhibitor ICRF-193,” Cell Cycle, Vol. 6, No. 10, 2007, pp. 1265-1267. doi:10.4161/cc.6.10.4225
- A. Zhang, Y. L. Lyu, C. P. Lin, N. Zhou, A. M. Azarova, L. M. Wood and L. F. Liu, “A Protease Pathway for the Repair of Topoisomerase II-DNA Covalent Complexes,” Journal of Biological Chemistry, Vol. 281, No. 47, 2006, pp. 35997-36003. doi:10.1074/jbc.M604149200
- H. Xiao, Y. Mao, S. D. Desai, N. Zhou, C. Y. Ting, J. Hwang and L. F. Liu, “The Topoisomerase IIbeta Circular Clamp Arrests Transcription and Signals a 26S Proteasome Pathway,” Proceedings of the National Academy of Sciences of the United States of America, Vol. 100, No. 6, 2003, pp. 3239-3244. doi:10.1073/pnas.0736401100
- W. P. Roos and B. Kaina, “DNA Damage-Induced Cell Death by Apoptosis,” Trends in Molecular Medicine, Vol. 12, No. 9, 2006, pp. 440-450. doi:10.1016/j.molmed.2006.07.007
- L. Wu, S. L. Davies, N. C. Levitt and I. D. Hickson, “Potential Role for the BLM Helicase in Recombinational Repair via a Conserved Interaction with RAD51,” Journal of Biological Chemistry, Vol. 276, No. 22, 2001, pp. 19375-19381. doi:10.1074/jbc.M009471200
- S. L. Ding, J. C. Yu, S. T. Chen, G. C. Hsu, S. J. Kuo, Y. H. Lin, P. E. Wu and C. Y. Shen, “Genetic Variants of BLM Interact with RAD51 to Increase Breast Cancer Susceptibility,” Carcinogenesis, Vol. 30, No. 1, 2009, pp. 43-49. doi:10.1093/carcin/bgn233
- O. S. Gildemeister, J. M. Sage and K. L. Knight, “Cellular Redistribution of Rad51 in Response to DNA Damage: Novel Role for Rad51C,” Journal of Biological Chemistry, Vol. 284, No. 46, 2009, pp. 31945-31952. doi:10.1074/jbc.M109.024646
- O. A. Hampton, P. Den Hollander, C. A. Miller, D. A. Delgado, J. Li, C. Coarfa, R. A. Harris, S. Richards, S. E. Scherer, D. M. Muzny, R. A. Gibbs, A. V. Lee and A. Milosavljevic, “A Sequence-Level Map of Chromosomal Breakpoints in the MCF-7 Breast Cancer Cell Line Yields Insights into the Evolution of a Cancer Genome,” Genome Research, Vol. 19, No. 2, 2009, pp. 167-177. doi:10.1101/gr.080259.108
- Y. Zheng, J. Zhang, K. Hope, Q. Niu, D. Huo and O. I. Olopade, “Screening RAD51C Nucleotide Alterations in Patients with a Family History of Breast and Ovarian Cancer,” Breast Cancer Research and Treatment, Vol. 124, No. 3, 2010, pp. 857-861. doi:10.1007/s10549-010-1095-5
- Y. Yarden and M. X. Sliwkowski, “Untangling the ErbB Signalling Network,” Nature Reviews Molecular Cell Biology, Vol. 2, No. 2, 2001, pp. 127-137. doi:10.1038/35052073
- A. Meindl, H. Hellebrand, C. Wiek, V. Erven, B. Wappenschmidt, D. Niederacher, M. Freund, P. Lichtner, L. Hartmann, H. Schaal, J. Ramser, E. Honisch, C. Kubisch, H. E. Wichmann, K. Kast, H. Deissler, C. Engel, B. Müller-Myhsok, K. Neveling, M. Kiechle, C. G. Mathew, D. Schindler, R. K. Schmutzler and H. Hanenberg, “Germline Mutations in Breast and Ovarian Cancer Pedigrees Establish RAD51C as a Human Cancer Susceptibility Gene,” Nature Genetics, Vol. 42, No. 5, 2010, pp. 410-414. doi:10.1038/ng.569
NOTES
*Corresponding author.