Advances in Aging Research
Vol.3 No.3(2014), Article ID:48288,13 pages
DOI:10.4236/aar.2014.33033
Correspondence amongst the PENO Test Battery Cognitive Results and Hippocampal Lesions in Alzheimer’s Disease
Rojas Karla1, Díaz Alfonso2, Espinosa Blanca3, Montaño Luis Felipe4, Dilhuydy Hugo5, Geraux Francine5, Joanette Yves5, Robitaille Yves6, Guevara Jorge7*
1Laboratorio Experimental de Enfermedades Neurodegenerativas, Instituto Nacional de Neurología y Neurocirugía “M.V.S.”, México DF, México
2Departamento de Farmacia, Facultad de Ciencias Químicas, BUAP, Puebla, México
3Departamento Bioquímica, INER, México DF, México
4Departamento de Biología Celular y Tisular, Facultad de Medicina, UNAM, México DF, México
5Centre de Recherche, Institut Universitaire de Gériatrie de Montréal, Montréal, Canadá
6Départament de Pathologie, Hôpital Ste-Justine, Montreal, Canadá
7Departamento de Bioquímica, Facultad de Medicina, UNAM, México DF, México
Email: *Jorge.Guevara@comunidad.unam.mx
Copyright © 2014 by authors and Scientific Research Publishing Inc.
This work is licensed under the Creative Commons Attribution International License (CC BY).
http://creativecommons.org/licenses/by/4.0/



Received 17 May 2014; revised 27 June 2014; accepted 25 July 2014
Alzheimer’s disease (AD) is characterized by a decline of cognitive functions. Distinctive histopathological hallmarks are neuritic plaques, neurofibrillary tangles, and synaptic alterations. Abnormally enlarged synaptic structures called “Meganeurite clusters” have been linked to plasticity changes. The aims of this study were to determine if cognitive impairment was related to specific neuritic and synaptic degeneration processes in patients with AD, and if the results of a cognitive test could be correlated with the histopathological damage. The neuropsychological evaluation obtained by the Protocole d’evaluation neuropsychologique optimal (PENO) test battery was used in live AD and control individuals. The histopathological evaluation of their brain after their death was carried out with specific polyclonal and monoclonal antibodies to Aβ, pTau protein, synaptophysin, and GAP-43. Images were obtained by confocal microscopy. The results showed a significant difference between healthy controls and Alzheimer’s patients in neuropsychological evaluation and histopathological hallmarks expression. The most significant positive correlation in AD patients was between memory and language results with the PENO test and the presence of Aβ +pTau+ plaques in the hippocampus. An interesting negative correlation was between cognitive impairment and the presence of Meganeuritic clusters, considered as “plasticity” markers. These results strongly supported the use of the PENO battery test to evaluate the progression of cognitive impairment in AD prone individuals and patients due to the strong correlation of the test results with histopathological brain lesions characteristic of Alzheimer’s disease.
Keywords:Cognitive Decline, Neuropsychological Evaluation, Synapsis, Plasticity, Alzheimer’s Disease

Alzheimer’s disease (AD) is a neurodegenerative and multifactorial illness [1] . The clinical symptoms involve cognitive impairment in different areas, mainly: memory, language, viso-constructive, spatial attention, praxis, gnosis, and executive functions [2] . When cognitive impairment is mild there is a believe that a short examination is sufficient to make the diagnosis, however, the most commonly used test for complaints of memory problems, the Minimental State Examination (MMSE) [3] , has low sensitivity for detecting mild cognitive impairment [4] . The Protocole d’evaluation neuropsychologique optimal (PENO) test battery [5] permits a superior neuropsychological evaluation in the progression of dementia; this test also brings to the fore distinct affected cognitive profiles (memory, language, praxis, gnosis and executive functions) in a transversal as well as longitudinal base, thus contributing to the early diagnosis of AD as it can differentiate between normal and pathological aging.
AD is characterized by the appearance of parenchymal amyloid-β deposits with and without neuritic elements, and by intraneuronal changes, including neurofibrillary tangles (NFTs) and synaptic loss [6] . Neuritic plaques comprise amyloid-β deposits surrounded by dystrophic neurites, reactive astrocytes, and microglia [7] [8] . Tau protein is the main component of NFTs and dystrophic neurites [9] [10] . Neurodegeneration in AD brains co-exists with a reactive plasticity event because Amyloid-β Deposits (AβDs), classic Neuritic Plaques (NPs), and Dystrophic Neurites Clusters lacking amyloid-β deposits (DNCs), all of which represent structures associated to the neuritic degenerative process, are co-located with reactive plasticity structures called Meganeuritic Clusters (MCs) and peri-neuronal sprouting in neurons of the hippocampus, which are strongly positive for synaptophysin and GAP-43 [11] [12] .
All mentioned above implies the co-participation of these neurodegenerative events in the distorted cognitive functions of AD patients. Recent studies have focused on identifying the beginning of the transition from healthy aging to dementia in order to identify the disease as early as possible [13] . Therefore the aim of this work was to determine a) if cognitive impairment was related to specific neuritic and synaptic degeneration processes in patients with AD, b) if the application of a cognitive test could serve early diagnostic purposes, and c) if a correlation between both could be found. We used the PENO test battery in a group of selected individuals, and after their deaths, we quantitated markers of neurodegeneration and reactive plasticity in their brains.
2.1. Subjects
The study was performed with data obtained from a sample of individuals recruited at the Institute Universitaire de Geriatrie de Montreal, all of which underwent neurological examination, neuroimaging studies, laboratory tests, computed tomography (CT) and/or magnetic resonance imaging (MRI) scans of the brain, neuropsychological testing and longuitudinal studies for cognitive deficits [5] . All the recruited individuals underwent recurrent six months evaluations using a validated neuropsychological battery. The Protocole d’evaluation neuropsychologique optimal (PENO) battery was used as clinical measure of global cognition [14] ; this battery evaluated logical memory, comprehension, word production, discursive abilities, visual discrimination, and bell test through 20 psychological tests [5] (Table 1). Highly trained research psychologists made the evaluations but for the purpose of our study, we included one final evaluation before the patient’s death. A diagnosis of dementia was made during a consensus meeting using DSM-IV criteria [15] . The neuropsychological evaluation of the control group was taken from a standardized tables result, depending on the patient’s age and education. APOE

Table 1 . Parameters evaluated by the protocole D’ evaluation neuropsychologique optimal (PENO) test.
genotype data were available but the individuals that were selected for this study were not selected based on their APOE genotypes. The clinical profile of the individuals includes bioethic, deontological, clinico-pathological, epidemiological, genetic, molecular, epidemiological, sociographic, toxic-environmental, biochemical, psychological, and neurological information. All patients and/or related family received full written and oral information regarding all study procedures and signed an informed consent. All experiments were done in accordance with the Helsinki Declaration of 1975 and the local ethics committee of the Institute de Geriatrie de Montreal, in accordance with current legislation on this field, approved the protocol as well as the details of the informed consent.
Brain tissue was obtained immediately after the death of non-traumatic origin of the patients. The study was conducted considering obtaining biopsies of 24 people: 11 individuals with cognitive decline-Alzheimer Diseases cases (mean age 76 ± 5.5 years old) and 13 non-demented controls (mean age 74.1 ± 6.9 years old) diagnosed clinically and histopathologically by two neuropathologists according to the NINCDS/RCDA criterion [16] [17] . Cortex tissue fragments ranging in size from 0.3 to 0.5 × 2 × 2 cm were obtained by an experimented technician within 24 hr after death and fixed by immersion in 4% paraformaldehyde for at least 36 hr. Eleven biopsy samples were acquired from the brain anatomical areas that correspond to the Brodmann’s areas described in Table 2 As part of the protocol biopsy samples from the eleven areas were stained to determine the expression of amyloid-β, phosphorylated tau protein and the presence of neurofibrillary tangles, neuritis dystrophy, and meganeuritic clusters. To quantitate the differences of the above mentioned markers only the results of brain anatomical areas 2, 4, 6 and 7 in both groups were considered.
2.2. Immunohistochemistry
Brain biopsies were paraffin embedded. Serial sections, 6-µm thick, were used for immunohistochemistry. Paraffin-embedded sections were dewaxed and rehydrated in decreasing concentrations of ethanol. When needed slides were pre-treated in 80% formic acid for 5 min to enhance amyloid-β immunoreactivity [11] [18] . Antigen retrieval was achieved by boiling the samples in 0.01 M sodium citrate pH 6 for 20 - 40 min. The quenching of the endogenous peroxidase was achieved by incubation with 0.3% hydrogen peroxide in methanol for 30 min at room temperature. After washing with phosphate-buffered saline (PBS) the slides were incubated with 1% albumin solution for 2 hr at room temperature, and extensively washed with PBS. The slides were incubated at 4˚C in a humid chamber overnight with primary antibody diluted in Tris-buffered saline containing 1% albumin. Mouse monoclonal anti-human hyper-phosphorylated PHF-tau (MABAD2) diluted 1:500 kindly provided by Dr. André Delacourte, mouse monoclonal anti-growth-associated protein 43 (MAB347) diluted 1:500, rabbit anti-neurofibrillary tangles (AB1518) antibody diluted 1:1000, and mouse monoclonal anti-synaptophysin (MAB332) diluted 1:1000, were from Millipore Co. (Billerica, MA). Rabbit anti-human tau (A002401) diluted 1:1000 was from Dako Co. (Carpinteria, CA).
Some biopsy slides were double stained to determine AD2 and amyloid-β co-expression. Briefly, sections were incubated overnight in a humidity chamber at 4˚C with the primary ant-AD2 antibody diluted 1:500 in PBS pH 7.4. After washing with PBS the slides were incubated for 1 hr at room temperature with a FITC-conjugated anti-isotype antibody (Jackson Immunoresearch, Canada). Samples were washed and incubated with a polyclonal anti-amyloid-β antibody (1:50, Boeringher-Manheim, Germany) for 2.5 hr at room temperature, washed with PBS and incubated for 1 hr at room temperature with a lissamine-rhodamine-conjugated anti-isotype antibody (Jackson Immunoresearch, Canada). Finally, the samples were washed and mounted with Vectashield (Vector laboratories, Burlingame).
Table 2. Location of biopsied brain regions.
2.3. Confocal Microscopy
Double-labeled slides were analyzed with a Zeiss microscope equipped with epi-ilumination and a dual laser confocal system (Zeiss LSM 410, Germany). Using a 40 × oil-immersion objective lens, consecutive images were captured in two channels (FITC A = 492; E = 520; lissamine-rhodamine A = 570; E = 590). The images were projected on the two-dimensional plane and were merged using a pseudocolor display (green for FITC and red for lissamine-rhodamine). In the merged image, a yellow color was interpreted as a co-localization between two fluorochromes.
Quantification of the number of plaques, neurofibrillary tangles and dystrophic neuritis: The density of pathological structures was evaluated using scans of all the tissue available on each sample, obtained with a 40X oil objective. Plaque subsets and subtypes were visually attributed according to their immunoreactivity. Since there were variations in the extent of tissue in the different sections, the subtype densities was normalized by dividing the number of pathological structures by the area of tissue. The gray matter area in each sample was determined by placing the slides under a CCD camera interfaced to an image analysis system (MCID Image Analysis System, Imaging Research Inc., ON. Canada); the density of plaques was automatically computed by image analysis software (IBAS, Kontron Electronik. Germany). After confocal analysis, total gray matter surface was reported as true density of pathological structures subsets/mm2.
Quantification of meganeuritic clusters. 11 cortical brain regions (in both hemispheres) were scanned using a 10× objective (total magnification = 100×). Counting the number of clusters on the three richest fields and then calculating an average value determined the density of the clusters. Total numbers of synaptophysin positive meganeuritic clusters were reported by mm2.
2.4. Statistical Analysis
Data were expressed as the mean ± SD of each group. We compared the neuropsychological evaluation and the mean value of the different markers whose expression was analyzed, using the Mann Whitney test. A p ≤ 0.05 was considered statistically significant. Due to the effect of some variables such as age, scholarity, income, etc. some results were normalized using the z-test (from −1 to +1) where the higher the punctuation the greater the deterioration. Analyses were carried out using the SPSS 13.0 software (Addinsoft; SPSS Inc., Chicago, IL, USA).
3.1. Neuropsychological Evaluation
All individuals asked to perform the test complied without difficulty. Neither educational status nor sensory disturbances such as mild visual or hearing disorders, limited the ability of the individuals to take the test. The time was spent to apply the test varied from 10 to 15 min. The results demonstrated significant differences in the cognitive function evaluated by the longitudinal studies battery for cognitive deficits between the control and the Alzheimer disease group. These results shown in Figure 1 corresponded to the normalized evaluation. The overall assessment of language ability that includes word comprehension, oral denomination, verbal fluency, discourse and reading abilities, and word repetition demonstrated significant differences between Alzheimer’s
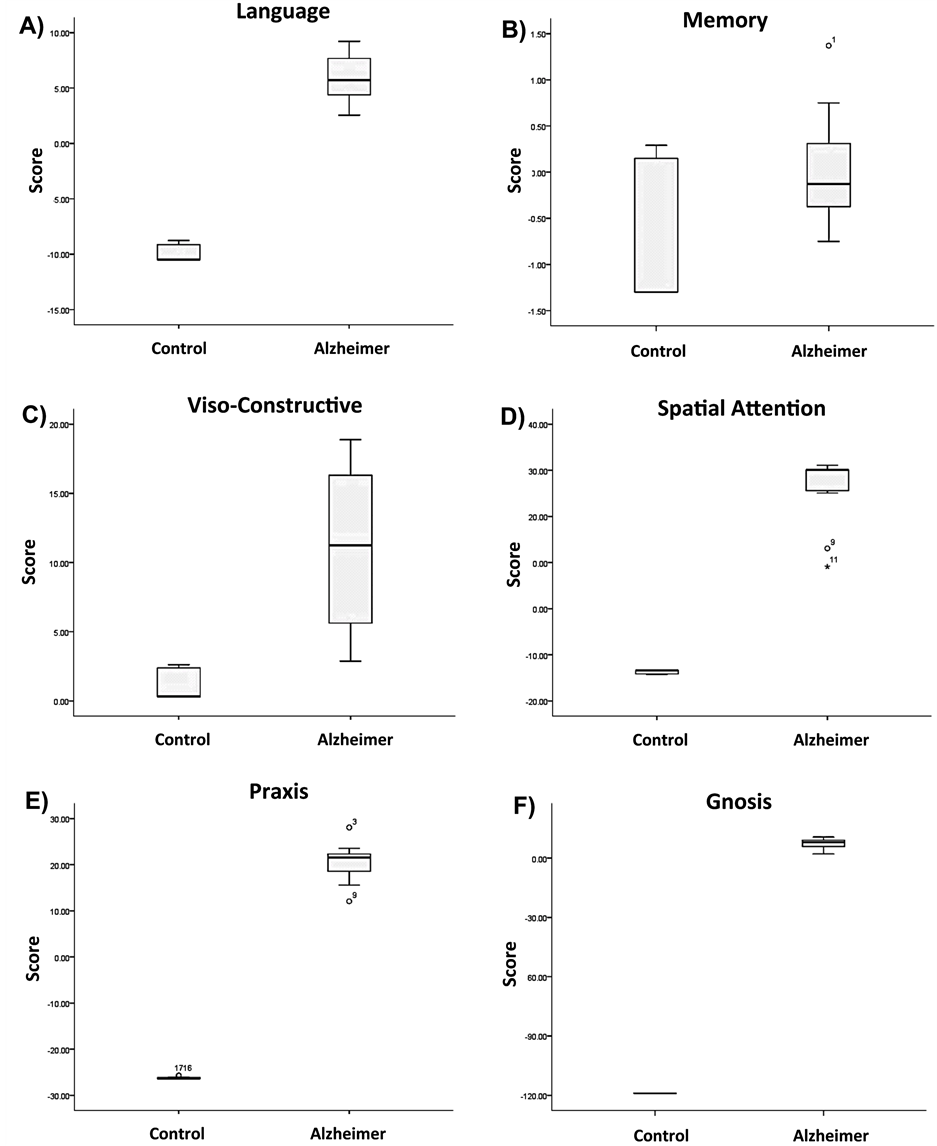
Figure 1. Correspond to the normalized evaluation box-plot of the last evaluation before the patient’s death. Mean ± ES.
individuals (mean 5.9 score vs. −9.8 in control individuals; p < 0.0001) (Figure 1(A)). The differences recorded and control in relation to memory (mean 0.01 vs. −0.62 score) (Figure 1(B)) indicated a decline in memory processes in the AD individuals but the difference was not statistical significant. The evaluation viso-constructive evaluation (mean 11.18 vs. 1.28 score) (Figure 1(C)) showed significant differences between groups (p < 0.001). The most evident and highly significant (p < 0.0005) deteriorations were in spatial attention (mean 25.7 vs. −13.7 score) (Figure 1(D)), praxis (mean 20.3 vs. −26.1 score) (Figure 1(E)) and gnosis (mean 7.39 vs. −118.9 score) (Figure 1(F)) in the AD group compared to the control group. These results clearly reveal that with the exception of memory patients with AD exhibit severe cognitive decline compared to the control group.
The evaluation of the same parameters with the PENO protocol considering non-normalized data uniquely from Alzheimer’s disease individuals showed interesting differences. PENO evaluation was only adjusted to age and schoolarity. Although the overall normalized PENO score was significantly higher in Alzheimer’s disease individuals than in the control individuals (Figure 2(A)) the results with non-normalized data showed that the
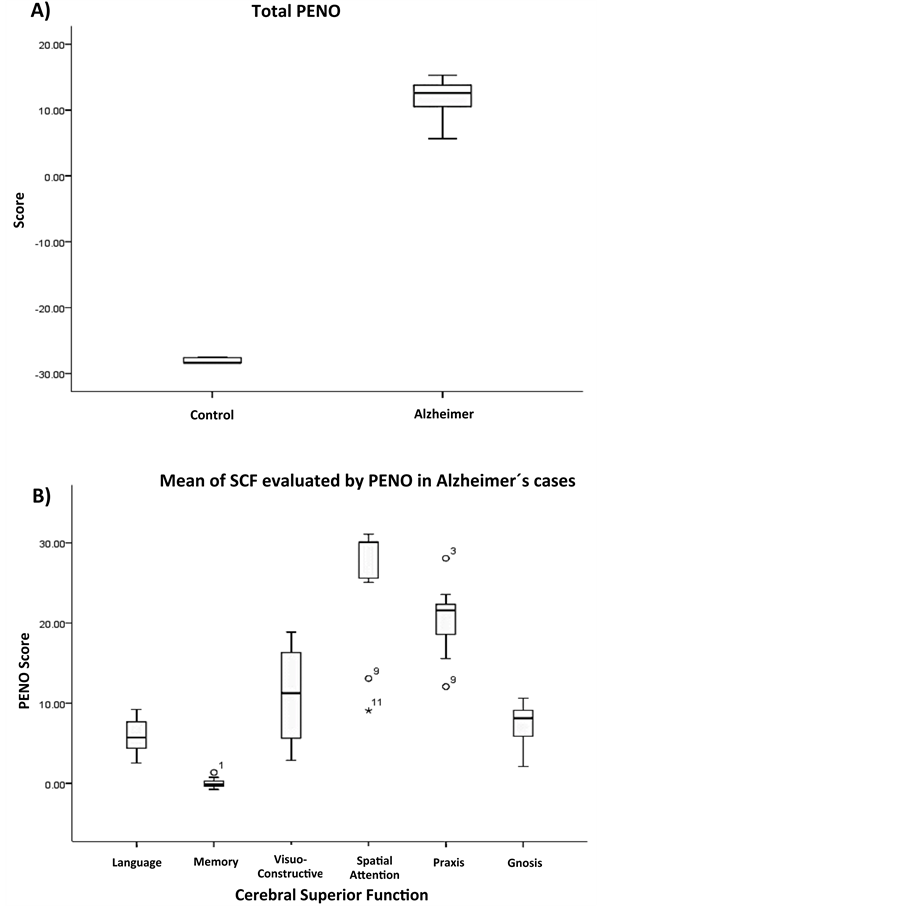
Figure 2. Box-plot showing A) the overall mean value of the PENO test results using non-normalized data; B) the mean cerebral superior function of Alzheimer’s disease patients considered the parameters determined by test. The results are expressed as mean ± SE.
less affected function was spatial attention whereas memory followed by gnosis were the most severely affected functions (Figure 2(B)). It was also interesting to observe the oscillatory pattern of cognitive decline, but as the figure shows some of the individuals did not conform to the pattern. Patient number 1 and 11 had Lewy bodies, patient 3 had an Alzheimer’s disease variant, and patient number 9 had severe vascular damage.
The quantification and comparison of histopathological hallmarks in the 2, 4, 6, and 7 anatomical areas of brain samples of the control and the Alzheimer’s disease group showed that the presence and quantitation of pathological structures were significantly higher in the Alzheimer disease group than in the control group. The only exception was the density of amyloid-β deposits in brain samples of individuals 60 years old or elder, independently of their status i.e., AD or control, although strangely, the density was lower in the AD samples (2.29 vs. 5.76, p = 0.777). Our most interesting result indicated a highly significant increase in Meganeuritic clusters density in Alzheimer’s disease (2.20 vs. 0.02, p < 0.001) but surprisingly there was an inverse and highly significant correlation between the neuropsychological evaluation results and the presence of meganeuritic clusters synaptophysin-reactive (Figure 3).
3.2. Neuropathology
The neurofibrillary degeneration recognized by the AD2 antibody and the amyloid-β deposits demonstrated six singular lesions in the brain biopsy slides: 1) amyloid-β deposits [diffuse senile plaques, SPs], 2) classic neuritic plaque with an amyloid-β core [NP], 3) clusters of amyloid-β-free dystrophic neurites [DNCs], 4) dystrophic neurites [DNs], 5) neurofibrillary tangles [Ab + pTau + plaques; NFT], and 6) meganeuritic clusters immunoreactive to synaptophysin [MCs]. The images shown in Figure 4 are representative of the above-mentioned lesions found in the hippocampus of AD brain biopsies. A further Spearman correlation analysis to identify whether two variables can relate to a monotonic function showed a positive correlation between density of meganeuritic clusters and PENO, and density of Ab + pTau + plaques with PENO. The other correlations gave negative coefficients (Figure 5).
Alzheimer disease is the most common age-related brain degeneration. The initial symptoms heralding Alzheimer’s disease are preceded by a pre-symptomatic stage characterized by destroyed synapses and connections [19] that provoke mild cognitive impairment and behavioral disturbances [20] . The search for simple diagnostic alternatives to detect early cognitive impairment is an active area of interest since many currently used or under evaluation choices lack specificity and/or are poor at quantitating damage. For instance, early image diagnosis is not very useful. For example, the early diagnosis of image is not very useful because, in Alzheimer’s disease, there is a deterioration in the medial temporal lobe, which is difficult to be identified and therefore, no image features are inconclusive [21] . The search for cerebrospinal fluid biomarkers has diagnostic potential [22] but its application as early diagnostic and follow-up tool is highly restricted. The detection of neural thread protein in urine samples has advantages [23] but very low protein concentrations and high salt levels make it difficult to use [24] . Molecular biomarkers such as members of the EGR family are associated to neuronal plasticity in the brain [25] but lack early diagnostic or progression usefulness.
Early cognitive impairment apparently involves Tau-protein-dependent lesions in selective brain regions. Tau is a protein that binds and stabilizes microtubules; when it remains phosphorylated, a hallmark of Alzheimer’s disease lesions [26] , destabilization of the microtubule and abnormal aggregation of tau protein to paired helical filaments occur, the results being neurofibrillary tangles (NFTs) deposition and eventually neuronal dead [27] [28] . Changes in cognition are associated with pretangle events within the cholinergic basal forebrain before frank NFT deposition [29] , or with plaques containing amyloid-β and phosphorylated tau protein [30] . The relative density and distribution of NFTs have been used to classify Alzheimer’s disease into three hypocampal subtypes that share slow cognitive decline [31] . NFTs, neuron loss and synaptic loss parallel the progression of cognitive decline [32] . Nevertheless, it is the number of NFTs and not the number of plaques, which correlate best with the degree of dementia in Alzheimer’s disease [9] [33] .
Normally cognition is evaluated by means of the Mini Mental State Examination but it is not considered a dementia sensitive test. Our results, using PENO as the last cognitive deficit test result of the patients before their death, showed that the most affected cerebral superior function was memory followed by language and gnosis, all of which are mainly associated with the hippocampus. Therefore biopsies specifically obtained from Brodman’s areas that define executive dysfunction (prefrontal cortex), apraxia (parietal cortex), visuospatial
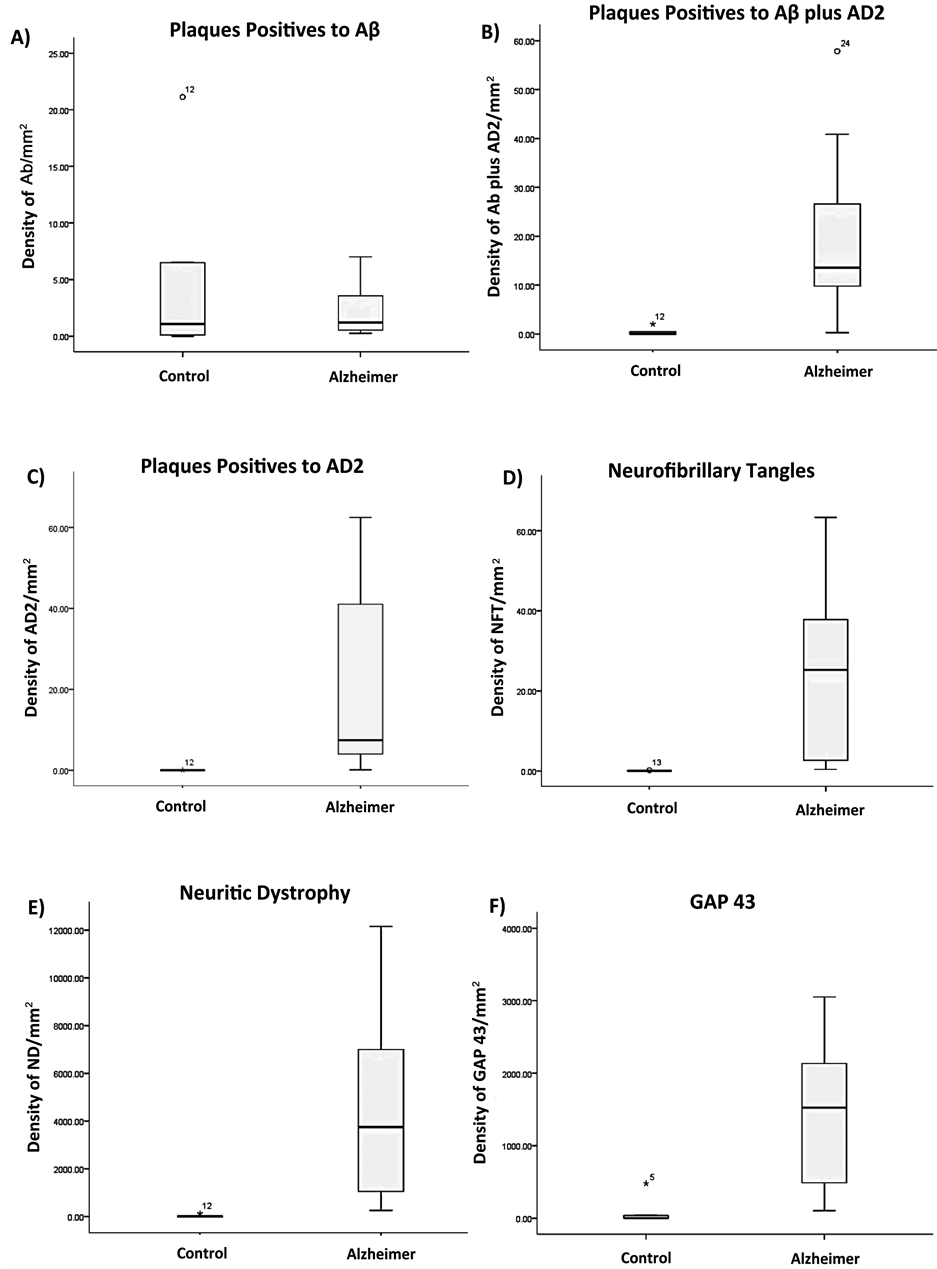
Figure 3. Histograms showing the results of the quantitation and correlations of Alzheimer’s disease hallmarks in the brain samples and the neuropsychological evaluations.
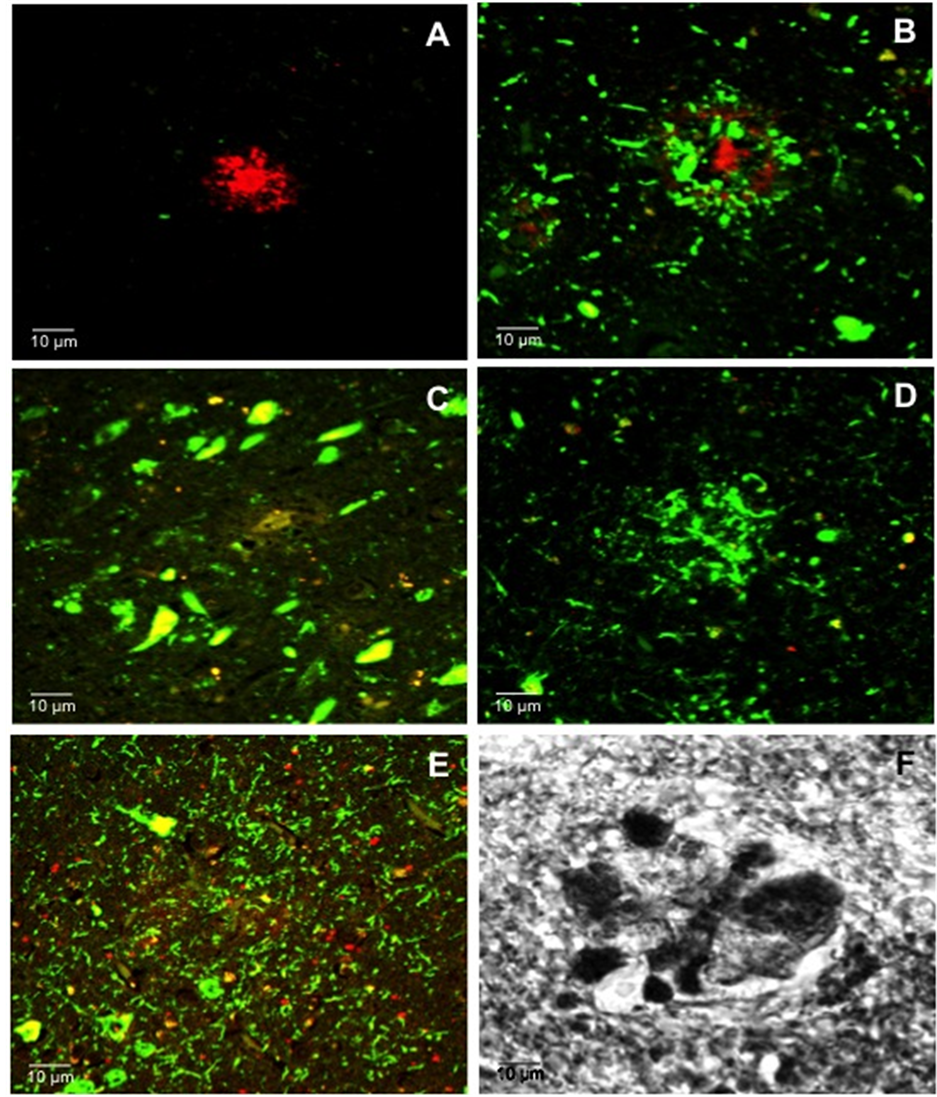
Figure 4. Representative confocal images of the main histological hallmarks found in the brain biopsies of Alzheimer’s disease patients. A) Aβ positive plaques; B) Neuritic plaque (Aβ and phosphorylated tau) plaque; C) Intracellular Neurofibrillary Tangles; D) Phosphorylated tau plaque; E) Distrofic neuritis positive to phosphorylated tau; F) Meganeuritic clusters positive to synaptophysin.
navigation deficit (occipito-parietal cortex), and semantic memory (anterior temporal cortex) were analyzed. We found that cognitive decline was associated with the presence of Aβ plaques containing phosphorylated tau, or Aβ negative plaques containing phosphorylated tau, classical neurofibrillary tangles, dystrophic neuritis, meganeuritic clusters which represent swollen terminal synapsis reactive to synaptophysin, and expression of neuronal
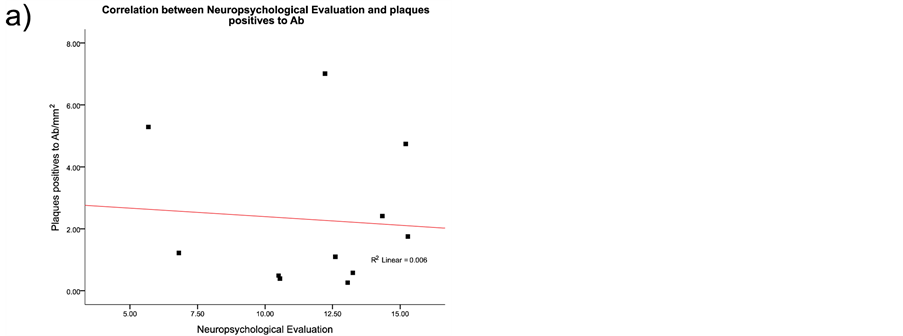
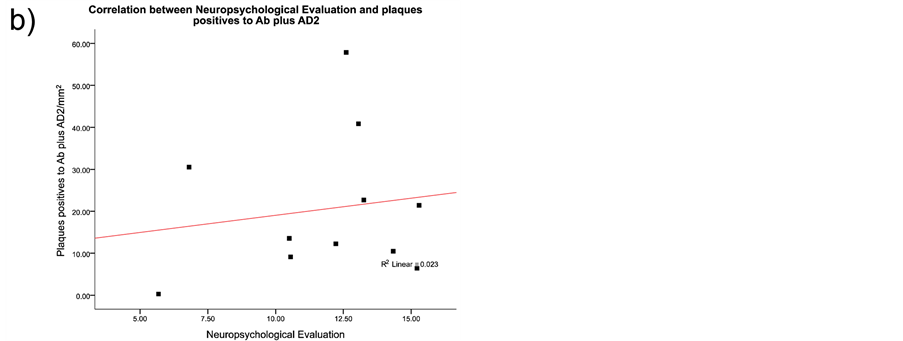
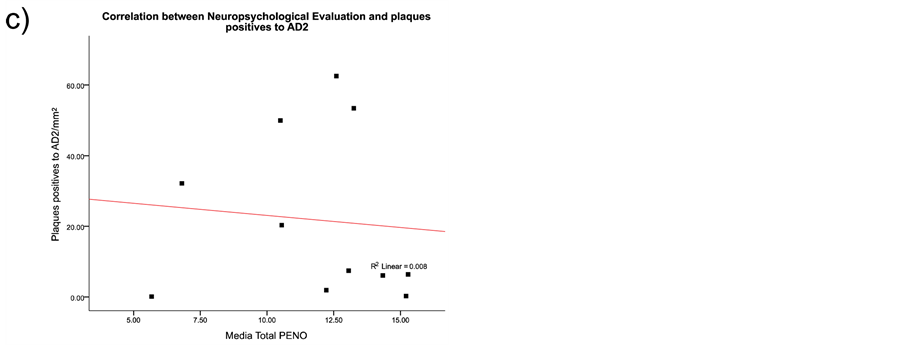
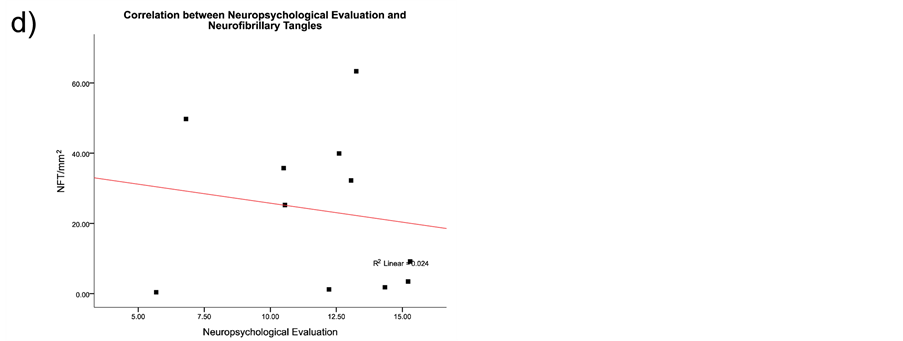
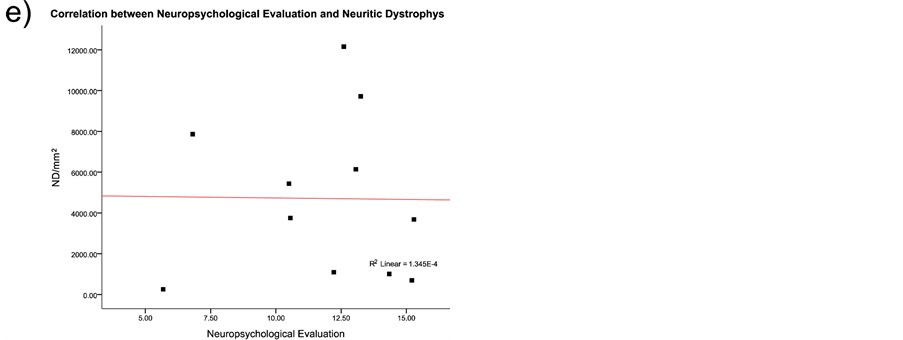
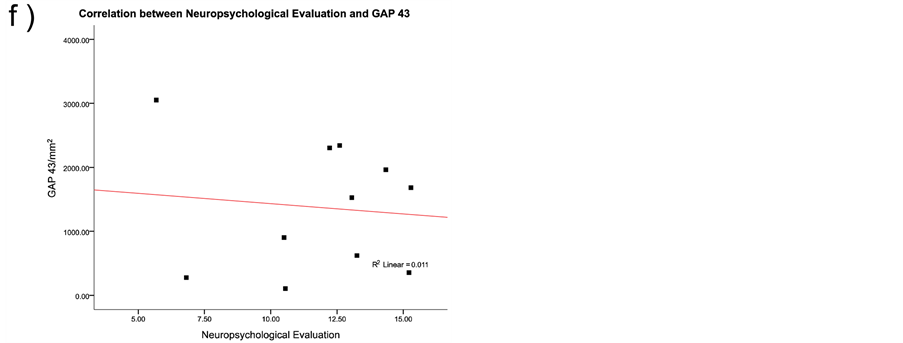
Figure 5. Spearman correlation results between the histological hallmarks and the neuropsychological evaluation in the Alzheimer’s disease patients. As discussed in the results section the only positive correlations were with decline in superior brain cognitive function and the presence of Aβ plus phosphorylated Tau protein plaques, or with the presence of meganeuritic clusters. Each dot represents the results of each Alzheimer’s disease patient.
growth-associated protein 43, an hippocampus phosphoprotein related to acquisition and processing of information [34] . Our results support the concept that Aβ plaques are normal in the aging process [35] [36] , but interestingly they also support the diagnostic usefulness of PENO test to detect early cognitive decline as the results of this battery showed a striking association with the presence of Aβ containing phosphorylated tau.
Synaptic damage and neuronal loss in AD could be counteracted by neuronal plasticity [37] . We found an inverse correlation between the number of meganeuritic cluster in the hippocampus and better cognitive performance. It is possible that the overexpression of synaptic proteins and increased dendritic branching could be a compensatory mechanism (plasticity) [18] against “synapse failure” in the Alzheimer brain [38] [39] , although there is strong evidence of reactive plasticity and neuron degeneration areas in the hippocampus of Alzheimer’s disease brains [12] . Nevertheless, the protective concept of brain reactivity plasticity, which arouse from the notion that educational level helps reduce the risk to develop AD [40] [41] , has been confirmed [42] . Therefore continuous brain activation should be always contemplated to diminish the risk of Alzheimer’s disease development.
Our results strongly support the use of the PENO battery test to evaluate the progression of cognitive impairment in AD prone individuals and patients due to the strong correlation of the test with histopathological brain lesions characteristic of Alzheimer’s disease.
Karla Rojas is a Ph.D. student in the Biological Sciences Postgraduate course, Universidad Nacional Autónoma de México, and a recipient of CONACyT-scholarship 203415.
The authors have no financial interests or any conflicts related to the material in the manuscript.
- Kotze, M.J. and Van Rensburg, S.J. (2012) Pathology Supported Genetic Testing and Treatment of Cardiovascular Disease in Middle Age for Prevention of Alzheimer’s Disease. Metabolic Brain Disease, 27, 255-266. http://dx.doi.org/10.1007/s11011-012-9296-8
- Slavin, M.J., Mattingley, J.B., Bradshaw, J.L. and Storey, E. (2002) Local-Global Processing in Alzheimer’s Disease: An Examination of Interference, Inhibition and Priming. Neuropsychologia, 40, 1173-1186. http://dx.doi.org/10.1016/S0028-3932(01)00225-1
- Folstein, M.F., Folstein, S.E. and McHugh, P.R. (1975) “Mini-Mental State”. A Practical Method for Grading the Cognitive State of Patients for the Clinician. Journal of Psychiatric Research, 12, 189-198. http://dx.doi.org/10.1016/0022-3956(75)90026-6
- Petersen, R.C., Smith, G.E., Waring, S.C., Ivnik, R.J., Tangalos, E.G. and Kokmen, E. (1999) Mild Cognitive Impairment: Clinical Characterization and Outcome. Archives of Neurology, 56, 303-308. http://dx.doi.org/10.1001/archneur.56.3.303
- Joanette, Y., Ska, B., Poissant, A., Belleville, S., Lecours, A.R. and Peretz, I. (1995) Evaluation neuropsychologique dans la demence de type alzheimers: Un Compromise Optimal. Lánné gérontogique, 9, 175-189.
- Terry, R.D., Masliah, E., Salmon, D.P., Butters, N., De Teresa, R., Hill, R., Hansen, L.A. and Katzman, R. (1991) Physical Basis of Cognitive Alterations in Alzheimer’s Disease: Synapse Loss Is the Major Correlate of Cognitive Impairment. Annals of Neurology, 30, 572-580. http://dx.doi.org/10.1002/ana.410300410
- Mark, R.J., Fuson, K.S. and May, P.C. (1999). Characterization of 8-Epiprostaglandin F2alpha as a Marker of Amyloid Beta-Peptide-Induced Oxidative Damage. Journal of Neurochemistry, 72, 1146-1153. http://dx.doi.org/10.1046/j.1471-4159.1999.0721146.x
- Maccioni, R.B., Farias, G., Morales, I. and Navarrete, L. (2010) The Revitalized Tau Hypothesis on Alzheimer’s Disease. Archives of Medical Research, 41, 226-231. http://dx.doi.org/10.1016/j.arcmed.2010.03.007
- Arriagada, P.V., Growdon, H.J., Hedley-Whyte, T. and Hyman, B.T. (1992) Neurofibrillary Tangles but Not Senile Plaques Parallel Duration and Severity of Alzheimer’s Disease. Neurology, 42, 631-639. http://dx.doi.org/10.1212/WNL.42.3.631
- Carter, M.D., Simms, G.A. and Weaver, D.F. (2010) The Development of News Therapeutics for Alzheimer’s Disease. Clinical Pharmacology & Therapeutics, 88, 475-486. http://dx.doi.org/10.1038/clpt.2010.165
- Espinosa, B., Zenteno, R., Mena, R., Robitaille, Y., Zenteno, E. and Guevara, J. (2001) O-Glycosylation in Sprouting Neurons in Alzheimer Disease, Indicating Reactive Plasticity. Journal of Neuropathology Experimental Neurology, 60, 441-448.
- Guevara, J., Dilhuydy, H., Espinosa, B., Delacourte, A., Quirion, R., Mena, R., Joanette, Y., Zenteno, E. and Robitaille, Y. (2004) Coexistence of Reactive Plasticity and Neurodegeneration in Alzheimer Diseased Brains. Histology and Histopathology, 19, 1075-1084.
- Johnson, D.K., Storandt, M., Morris, J.C. and Galvin, J.E. (2009) Longuitudinal Study of the Transition from Healthy Aging to Alzheimer Disease. Archives of Neurology, 66, 1254-1259. http://dx.doi.org/10.1001/archneurol.2009.158
- Joanette, Y., Poissant, A., Ska, B. and Fontaine, F. (1990) Protocole D’Evaluation Neuropsychologique Optimal (PENO). Montreal: Laboratoire Theophile-Alajouanine, Centre de recherché du Centre hospitalier Cótes-des-Neiges.
- Frances, A., Mack, A.H., First, M.B., Widiger, T.A., Ross, R., Forman, L. and Davis, W.W. (1994) DMS-IV Meets Philosophy. Journal of Medicine and Philosophy, 19, 207-218. http://dx.doi.org/10.1093/jmp/19.3.207
- Khachaturian, Z.S. (1985). Diagnosis of Alzheimer’s Disease. Archives of Neurology, 42, 1097-1105. http://dx.doi.org/10.1001/archneur.1985.04060100083029
- Mirra, S.S., Heyman, A., McKeel, D., Sumi, S.M., Crain, B.J., Brownlee, L.M., Vogel, F.S., Hughes, J.P., van Belle, G. and Berg, L. (1991) The Consortium to Establish a Registry for Alzheimer’s Disease (CERAD). Part II. Standarization of the Neuropathologic Assessment of Alzheimer’s Disease. Neurology, 41, 479-486. http://dx.doi.org/10.1212/WNL.41.4.479
- Guevara, J., Espinosa, B., Zenteno, E., Vazquez, L., Luna, J., Perry, G. and Mena, R. (1998) Altered Glycosylation Pattern of Proteins in Alzheimer Disease. Journal of Neuropathology & Experimental Neurology, 57, 905-914. http://dx.doi.org/10.1097/00005072-199810000-00003
- D’Amelio, M. and Rossini, P.M. (2012) Brain Excitability and Connectivity of Neuronal Assemblies in Alzheimer’s Disease: From Animal Models to Human Findings. Progress in Neurobiology, 99, 42-60. http://dx.doi.org/10.1016/j.pneurobio.2012.07.001
- Petersen, R.C., Doody, R., Kurz, A., Mohs, R.C., Morris, J.C., Rabins, P.V., Ritchie, K., Rossor, M., Thal L. and Winblad, B. (2001) Current Concepts in Mild Cognitive Impairment. Archives of Neurology, 58, 1985-1992. http://dx.doi.org/10.1001/archneur.58.12.1985
- Zeineh, M.M., Holdsworth, S., Skare, S., Atlas, S.W. and Bammer, R. (2012) Ultra-High Resolution Diffusion Tensor Imaging of the Microscopic Pathways of the Medial Temporal Lobe. Neuroimage, 62, 2065-2082. http://dx.doi.org/10.1016/j.neuroimage.2012.05.065
- Anoop, A., Singh, P.K., Jacob, R.S. and Maji, S.K. (2010) CSF Biomarkers for Alzheimer’s Disease Diagnosis. International Journal of Alzheimer’s Disease, 2010, 1-12. http://dx.doi.org/10.4061/2010/606802
- Levy, S., McConville, M., Lazaro, G.A. and Averback, P. (2007) Competitive ELISA Studies of Neural Thread Protein in Urine in Alzheimer’s disease. Journal of Clinical Laboratory Analysis, 21, 24-33. http://dx.doi.org/10.1002/jcla.20159
- Thongboonkerd, V. (2007) Practical Points in Urinary Proteomics. Journal of Proteome Research, 6, 3881-3890. http://dx.doi.org/10.1021/pr070328s
- Gómez-Ravetti, M., Rosso, O.A., Berretta, R. and Moscato, P. (2010). Uncovering Molecular Biomarkers That Correlate Cognitive Decline with the Changes of Hippocampus Gene Expression Profiles in Alzheimer’s Disease. PLoS One, e10153. http://dx.doi.org/10.1371/journal.pone.0010153
- Iqbal, K. and Grundke-Iqbal, I. (2008) Alzheimer Neurofibrillary Degeneration: Significance, Etiopathogenesis, Therapeutics and Prevention. Journal of Cellular and Molecular Medicine, 12, 38-55. http://dx.doi.org/10.1111/j.1582-4934.2008.00225.x
- Matenia, D. and Mandelkow, E.M. (2009) The Tau of MARK: A Polarized View of the Cytoskeleton. Trends in Biochemical Sciences, 34, 332-342. http://dx.doi.org/10.1016/j.tibs.2009.03.008
- Kosik, K.S. (1993) The Molecular and Cellular Biology of Tau. Brain Pathology, 3, 39-43. http://dx.doi.org/10.1111/j.1750-3639.1993.tb00724.x
- Vana, L., Kanaan, N.M., Uqwu, I.C., Wuu, J., Mufson, E.J. and Binder, L.I. (2011) Progression of Tau Pathology in Cholinergic Basal Forebrain Neurons in Mild Cognitive Impairment and Alzheimer’s Disease. American Journal of Pathology, 179, 2533-2550. http://dx.doi.org/10.1016/j.ajpath.2011.07.044
- Price, J.L., McKeel Jr., D.W., Buckles, V.D., Roe, C.M., Xiong, C., Grundman, M., Hansen, L.A., Petersen, R.C., Parisi, J.E., Dickson, D.W., Smith, C.D., Davis, D.G., Schmitt, F.A., Markesbery, W.R., Kaye, J., Kurlan, R., Hulette, C., Kurland, B.F., Higdon, R., Kukull, W. and Morris, J.C. (2009) Neuropathology of Nondemented Aging: Presumptive Evidence for Preclinical Alzheimer Disease. Neurobiology of Aging, 30, 1026-1036. http://dx.doi.org/10.1016/j.neurobiolaging.2009.04.002
- Janocko, N.J., Brodersen, K.A., Soto-Ortolaza, A.I., Ross, O.A., Liesinger, A.M., Duara, R., Graff-Radford, N.R., Dickson, D.W. and Murray, M.E. (2012) Neuropathologically Defined Subtypes of Alzheimer’s Disease Differ Significantly from Neurofibrillary Tangle-Predominant Dementia. Acta Neuropathologica, 124, 681-692. http://dx.doi.org/10.1007/s00401-012-1044-y
- Serrano-Pozo, A., Frosch, M.P., Masliah, E. and Hyman, B.T. (2011) Neuropathological Alterations in Alzheimer Disease. Cold Spring Harbor Perspectives in Medicine, 1, 1-23. http://dx.doi.org/10.1101/cshperspect.a006189
- Alafuzoff, I., Iqbal, K., Friden, H., Adolfsson, R. and Winblad, B. (1987) Histopathological Criteria for Progressive Dementia Disorders: Clinical-Pathological Correlation and Classification by Multivariate Data Analysis. Acta Neuropathologica, 74, 209-225. http://dx.doi.org/10.1007/BF00688184
- Nelson, R.B., Friedman, D.P., O’Neill, J.B., Mishkin, M. and Routtenberg, A. (1987) Gradients of Protein Kinase C Substrate Phosphorylation in Primate Visual System Peak in Visual Memory Storage Areas. Brain Research, 416, 387-392. http://dx.doi.org/10.1016/0006-8993(87)90924-3
- Davies, D.C. and Hardy, J.A. (1988) Blood Brain Barrier in Ageing and Alzheimer’s Disease. Neurobiology of Aging, 9, 46-48. http://dx.doi.org/10.1016/S0197-4580(88)80017-4
- Delàere, P., Duyckaerts, C., Masters, C., Beyreuther, K., Piette, F. and Hauw, J.J. (1990) Large Amounts of Neocortical Beta A4 Depositis without Neuritic Plaques nor Tangles in a Psychometrically Assessed, Non-Demented Person. Neuroscience Letters, 116, 87-93. http://dx.doi.org/10.1016/0304-3940(90)90391-L
- Spires-Jones, T. and Knafo, S. (2012) Spines, Plasticity, and Cognition in Alzheimer’s Model Mice. Neural Plasticity, 2012, Article ID: 319836. http://dx.doi.org/10.1155/2012/319836
- Haass, C. and Selkoe, D.J. (2007) Soluble Protein Oligomers in Neurodegeneration: Lessons from the Alzheimer’s Amyloid β-Peptide. Nature Reviews Molecular Cell Biology, 8, 101-112. http://dx.doi.org/10.1038/nrm2101
- Sheng, M., Sabatini, B.L. and Sudhof, T.C. (2012) Synapses and Alzheimer’s Disease. Cold Spring Harbor Perspectives in Biology, 4, 1-18. http://dx.doi.org/10.1101/cshperspect.a005777
- Gatz, M., Mortimer, J.A., Fratiglioni, L., Johansson, B., Berg, S., Andel, R., Crowe, M., Fiske, A., Reynolds, C.A. and Pedersen, N.L. (2007) Accounting for the Relationship between Low Education and Dementia: A Twin Study. Physiology & Behavior, 92, 232-237. http://dx.doi.org/10.1016/j.physbeh.2007.05.042
- Mortimer, J., Fortier, I., Rajaram, L. and Gauvreau, D. (1998) Higher Education and Socioeconomic Status in Childhood Protect Individuals at Genetic Risk of AD from Expressing Symptoms in Late Life: The Saguenay-Lac-Saint-Jean Health and Aging Study. Neurobiology of Aging, 19, S215.
- Sjostrom, P.J., Rancz, E.A., Roth, A. and Hausser, M. (2008) Dendritic Excitability and Synaptic Plasticity. Physiological Reviews, 88, 769-840. http://dx.doi.org/10.1152/physrev.00016.2007

*Corresponding author.