Neuroscience & Medicine
Vol.2 No.2(2011), Article ID:5520,11 pages DOI:10.4236/nm.2011.22014
Advances in Oligoprotection
![]()
National Centre for Drug Screening, China Pharmaceutical University, Nanjing, China.
Email: liaohong56@hotmail.com
Received April 6th, 2011; revised April 17th, 2011; accepted May 9th, 2011.
Keywords: Oligodendrocytes, Neurovascular Unit, Oxidative Stress, Excitotoxicity, Demyelinating Diseases, Stroke, Spinal Cord Injury, Neuropharmacology
ABSTRACT
Oligodendrocytes, the myelinating glial cells of the nervous system, in various disease states display great vulnerability to excitotoxic damage, oxidative stress, and inflammatory cytokines. Besides demyelinating diseases where oligodendrocyte injury is primarily implicated, damage to them also occurs secondarily in various neuropathies. Oligoprotection should, therefore, be looked into something not just as a means for protecting oligodendrocytes alone but also as a common target of protecting neurons and the neurogliovascular unit as a whole. In this review, we provide a description on oligodendrocytes, the reasons for their vulnerability, the evolving new concepts of protecting neurovascular unit and recently neurogliovascular unit; the various diseases where oligodendrocyte injury is implicated with a brief idea on different injury mechanisms of oligodendrocytes. Finally, we present the summary of the drugs that have shown promising results in protecting oligodendrocytes or in protecting white matter as a whole in different in vivo and in vitro models.
1. Introduction
Neurobiology has been a field of ever-growing interest with more and more researchers working aimed at preventing or curing various neurological diseases like Multiple Sclerosis (MS), Alzheimer’s disease (AD), Parkinson’s disease (PD). The incidence of these diseases is on the rise with the increasing elderly population. Our high paced life style has increased auto-accidents and the cases of traumatic brain injury (TBI) and spinal cord injury (SCI) which leads to loss of lives or debilitating conditions lifelong.
Research focused in central nervous system (CNS) pathologies in the yesteryears focused centrally on neuronal mechanisms. However, the concept of neuroprotection alone failed to prove optimal therapeutic strategy hence the neuron-based model has taken a paradigm shift to integrate the non-neuronal cells which maintain the homeostatic environment for neurons. It has widened to embrace much wider concept of Neurovascular unit (NVU) which itself has remolded into Neurogliovascular unit [1,2]. The neurovascular unit has been recently defined as a unit that comprises neurons and non-neuronal cells such as vascular cells (endothelia, pericytes and vascular smooth muscle cells) and glia (astrocytes, microglia and oligodendrocytes), along with an extracellular matrix that maintains the integrity of brain tissue [3]. Also the concept of oligovascular niche has emerged which includes the main cell types comprising white matter—the neuronal axon, oligodendrocyte (myelin), astrocyte, and endothelial cell [1].
Oligodendrocytes (OLGs) are the myelinating glial cells of the CNS. OLGs in the nervous system are analogous to the Schwann cells in the peripheral nervous system in providing support for quick transfer of neuronal signals. OLGs not only myelinate axons but also maintain their functional integrity and survival thorough oligodendrocyte specific proteins and/or trophic factor release by secreting soluble factors. Factors like Brain derived neurotrophic factor (BDNF), Neurotrophin-3 (NT-3), Nerve growth factor (NGF), and Platelet derived growth factor (PDGF) are reported as oligodendrocyteaxon signaling mediators [1]. They differ greatly morphologically based on their function and subtypes [4].
Myelin sheath, the protective covering of the nerve fibers insulating the axons, is responsible for transmission of the electrical signals with high velocity and without the loss of signal strength. The myelin sheath produced by OLGs is up to 600 × surface area and 100 × weight of soma and therefore their energy requirement is 2-3 times more than other brain cells [5]. These facts make OLGs the most vulnerable cells in the brain coupled with highest rate of metabolism, high reactive oxygen species (ROS) formation, low levels of antioxidant glutathione; high iron stores for cholesterol and lipid synthesis required for myelin formation; high levels of polyunsaturated fatty acids (PUFA) which react with ROS triggering lipid peroxidation [5-7]. OLGs are damaged as a result of adverse environmental signals and stress situations, such as oxidation, which may retard or inhibit myelination [8]. Neurons require assistance of glial cells for maintaining brain homeostasis from extracellular concentrations of ions, neurotransmitters and neurohormones for provision of an uninterrupted metabolic support [9]. The denuding of axons following oligodendrocyte death leads to functional loss of neurons and eventually their death, moreover, with a single oligodendrocyte myelinating numerous axons, death of a single oligodendrocyte exposes multiple neurons to risk [9].
2. Oligodendrocytes in Disease States
2.1. Multiple Sclerosis (MS)
MS is an autoimmune demyelinating disease with a neurodegenerative component most commonly diagnosed in patients 20 - 40 years of age [10]. In MS, the immune system attacks the white matter of the brain and spinal cord, causing multifocal demyelination leading to disability and/or paralysis. In acute MS, oligodendrocytes die mainly by apoptosis, however, in chronic MS lesion apoptosis was absent or rare which could because of the rapid removal of apoptotic cells in vivo [11]. Oxidative stress plays an important role in oligodendroglial death in MS [12]. Macrophages from MS patients show increased production of superoxide radicals and H2O2; while CSF displays significantly high level of NO metabolites, nitrite and nitrate compared to controls [13]. As the MS lesion formation starts, locally produced ROS induce tight junction and cytoskeletal changes in brain endothelial cells which facilitate transendothelial monocyte migration [6]. ROS also has a critical role in myelin phagocytosis and it has been observed that using ROS inhibitors like NADPH oxidase inhibitors, lipoic acid or catalase enzyme reduced myelin phagocytosis by macrophages [6]. Excitatory glutamate levels are increased in acute MS lesions and in normal-appearing white matter in MS patients [14]. In addition, oxidative stress contributes to the increase in glutamate concentrations in the extracellular space, as free radicals reduce the efficiency of GluTs [14]. TNF-α is found at high levels in multiple sclerosis (MS) plaques and induces both apoptotic and necrotic death of OLGs and inhibition of caspase-1 and caspase-3 protects them against TNF-α-induced apoptosis [11]. Interferon gamma (IFN-γ) produced by lymphocytes in MS lesions plays a role in apoptosis of oligodendrcoytes both in vivo and in vitro [11]. TNF-α increases the IFN-γ-induced death of OPCs and this effect can be partially suppressed by caspase inhibition [11]. Interleukin-11 is expressed by reactive astrocytes in MS tissue samples along the myelinated border of both active and silent lesions [15].
2.2. Paraventricular Leukomalacia (PVL)
Paraventricular leukomalacia (PVL) is the leading cause of brain damage in preterm infants, prior to 32 weeks of gestation; resulting in cerebral palsy and cognitive impairment [16,17]; and occurs due to damage to OPCs and hypomyelination [18]. It involves death of small areas of brain tissue around ventricles, the fluid filled areas of brain; creating holes in the brain [19]. The pathogenesis of OPC injury relates to hypoxia-ischemia and systemic infection/inflammation, common occurrences in premature infants. Three downstream mechanisms have been identified, i.e., microglial activation, excitotoxicity and free radical attack [20].
2.3. Stroke
Stroke, a leading cause of death occurs due to the interruption of blood supply to any part of the brain owing to the blockade or bursting of blood vessels, hence classified as ischemic and hemorrhagic stroke [21]. Immediately following interrupted blood supply to brain, the injury process initiates, if recovery of normal blood flow is not achieved within a short period of time, death of cells within the ischemic core begins [22]. Stroke disrupts the blood brain barrier (BBB) which in turn affects stroke progression and neuroregeneration [23]. Ischemia, in stroke, is known to cause cell death mainly through the oxidative stress and excitotoxicity. Energy deprivation in stroke leads to neuronal death, axonal dysfunction and loss of OLGs which are very sensitive to transient oxygen and glucose deprivation. It has been seen that OLGs in immature state are even more sensitive to ischemic injury in mature forms. These cells may release glutamate under ischemic conditions by reverse functioning of their glutamate transporter (GluTs) [14]. During ischemia/ reperfusion of brain tissue, oxidative stress sets in with a rise in Hydrogen Peroxide (H2O2) levels for about 1hr reaching up to 175 μM (approx.) [14]. Also, increased TNF-mRNA is seen 3 hr after stroke in transient Middle cerebral artery occlusion (MCAO) models in rats [23].
In humans, almost all cases of ischemic stroke involve white matter [24]. White matter ischemia may activate proteases like calpains which degrade neurofilament and cause phosphorylation of axonal microtubule proteins like tau. Thereby, they weaken the structural integrity of axons and also impair axonal mechanisms for anterograde and retrograde transport [25].
2.4. Spinal Cord Injury (SCI)
Spinal cord injury (SCI) refers to the damage to the spinal cord resulting in permanent neurological deficit. It is caused by direct injury or indirectly from damage to the surrounding bones, tissues, or blood vessels resulting from auto accidents, falls, sports injuries and other accidents. It is a very tragic case with higher prevalence in young and otherwise healthy individuals, thus creating physical, emotional, and economic burdens on both sufferer and society [26]. The loss of axonal transmission of sensory and motor nerves occurs distal to point of SCI, leading to multiple health problems such as recurrent kidney stones, urinary tract infection, pressure sores, and cardiac and respiratory dysfunction [27]. Acute SCI involves mechanical primary and biochemical secondary mechanism of injury; primary due to blunt cord compression owing to dislocation of spinal cord or bone fragments; biochemical factors in hemorrhagic necrotic tissue causing secondary damage to spinal cord [28]. SCI causes strong up-regulation of genes involved in transcription, inflammation and cell death [29]. Excitotoxicity, oxidative stress, lipid peroxidation/free radical injury, inflammatory factors all seem to play parts worsening the homeostasis of the brain and causing cell death [28].
2.5. Leukodystrophy
Leukodystrophies are rare diseases appearing during infancy or childhood and characterized by a progressive degeneration of the white matter due to imperfect growth or development of the myelin sheath [30]. Myelin is a complex framework made up of at least ten different chemicals. The leukodystrophies are a group of disorders caused by genetic defects in how myelin produces or metabolizes its chemicals components [4]. Each type of leukodystrophy results from the defect in the gene that controls one of the chemicals causing defects in enzymes such as arylsulphatase A (Metachromatic Leukodystrophy), aspartoacylase (Canavan disease), galactosylceramidase (Krabbe disease) or in enzymes responsible for breaking down phytanic acid (Refsum’s disease), defects in a fatty acid transfer protein (Adrenoleukodystrophy), or defects in the myelin protein PLP1 (Pelizaeus-Merzbacher disease) [31].
2.6. Alzheimer’s Disease (AD)
AD is a neurodegenerative disease with stealth onset that progresses relatively rapidly with the parenchymal deposition of beta amyloid (Aβ) in the form of senile/neuritic plagues and the neurofibrillatory tangles (NFT) resulting in the loss of neurons and synapses [32]. The white matter lesions and myelin abnormalities are common in the AD patients [33]. A is known to induce apoptosis of mature OLGs which can be attenuated by antioxidants. A also causes oxidative stress by upregulation of inducible nitric oxide synthase (iNOS) in mature OLGs through a TNF-α-mediated ceramide pathway [32]. Some cases of AD are caused by mutation of presenilin-1 gene. It has been seen that glutamate-mediated excitotoxicity in OLGs is potentiated by presenilin-1 mutation [31]. OLGs that differentiate and myelinate later in life produce thinner myelin sheaths around axons and these neurons have been observed to be more susceptible to the damage in AD [32]. The transentorrhinal region and nearby neocortical association areas are the late-myelinating regions known; these areas are also the seat for early Aβ deposits and myelin breakdown [5].
2.7. Schizophrenia (SZ)
Schizophrenia is a complex mental disorder of unknown etiology, usually surfacing in the young adulthood, which carries a genetic link [34]. Neuroimaging and microarray studies have provided evidence for myelin and oligodendrocyte abnormalities in SZ. As evidenced from electron microscopy, OLGs are the most severely affected cells in SZ undergoing dystrophy, necrosis and apoptosis [35]. Decreased expression of myelin and oligodendrocyterelated genes is seen in postmortem samples obtained from prefrontal cortex regions of schizophrenic patients, suggesting OLG loss or disruption at functional levels [36]. Six myelin-related genes specific to OLGs have decreased expression levels in schizophrenia. OLGs are decreased in number in the superior frontal gyrus of schizophrenics. Absence of myelin associated glycoprotein (MAG) genes may be the link in schizophrenia since MAG-knockout mice develops as a mice model for the disease [37]. Abnormal glutamate homeostasis is considered one of the leading pathological mechanisms of schizophrenia and this also puts OLGs at increased risk for excitototic damage [38]. Abnormalities in the white matter have been observed; however, so far it is not clear whether it is a cause or a consequence of the disease [32].
2.8. Other Diseases
Oligodendrocytes are also injured in a variety of other diseases like Progressive Multifocal Leukoencephalopathy [7], Post-Infectious Encephalitis [39], and also following radiation injury [40], etc.
3. Different Mechanisms of Injury to Oligodendrocytes
Oligodendrocytes known to be the most vulnerable cells in the brain undergo injury in different disease states by various mechanisms like oxidative stress, excitotoxicity, and inflammatory cytokines. Developing therapeutic approaches to OLG injuries and related diseases needs better understanding of the underlying mechanism by which damage occurs to these cells. The mechanism of injury in neurons which has been researched for long may not fit in case of OLGs and hence should be considered separately. The injury mechanism and mediators causing the injury in disease states are often intertwined with one type eliciting the other in response. Various mechanisms of injury to OLGs has been summed up and presented below in Figure 1.
3.1. Oxidative Stress
OLGs on exposure to reactive oxygen species (ROS) causes formation of lipid peroxides which further react with proteins leading to oxidative modifications of amino acid residue. Also the protein aggregates are formed and protein fragmentation occurs [41], and also leads to DNA fragmentation and finally apoptosis of cells. H2O2, a ma jor ROS, is normally produced in the brain in small amounts. Its toxicity is due to its reaction with transition metals (iron-mediated Fenton reaction) to form hydroxyl radicals, which can further react with amino acids, nucleic acids, phospholipids and sugars, eventually causing cell death [42].
3.2. Excitotoxicity
Oligodendrocytes injury and death occurs on exposure to excitotoxic agents like glutamate and extraceullular ATP [14]. Mature OLGs undergo necrosis on exposure to kainite through the Alpha-amino-3-hydroxy-5-methyl-4- isoxazolepropionic acid (AMPA), and kainite (KA) receptors; recently N-methyl-D-aspartate (NMDA) have also been known to play role in OLG pathology [43,44]. Oka et al. was the first to provide evidence that OLGs are highly vulner able to glutamate using the primary cultures. The oligodendroglial toxicity is mediated by a transporter-related mechanism involving the inhibition of cystine uptake, which results in glutathione depletion and cellular vulnerability to toxic-free radicals. Glutamate
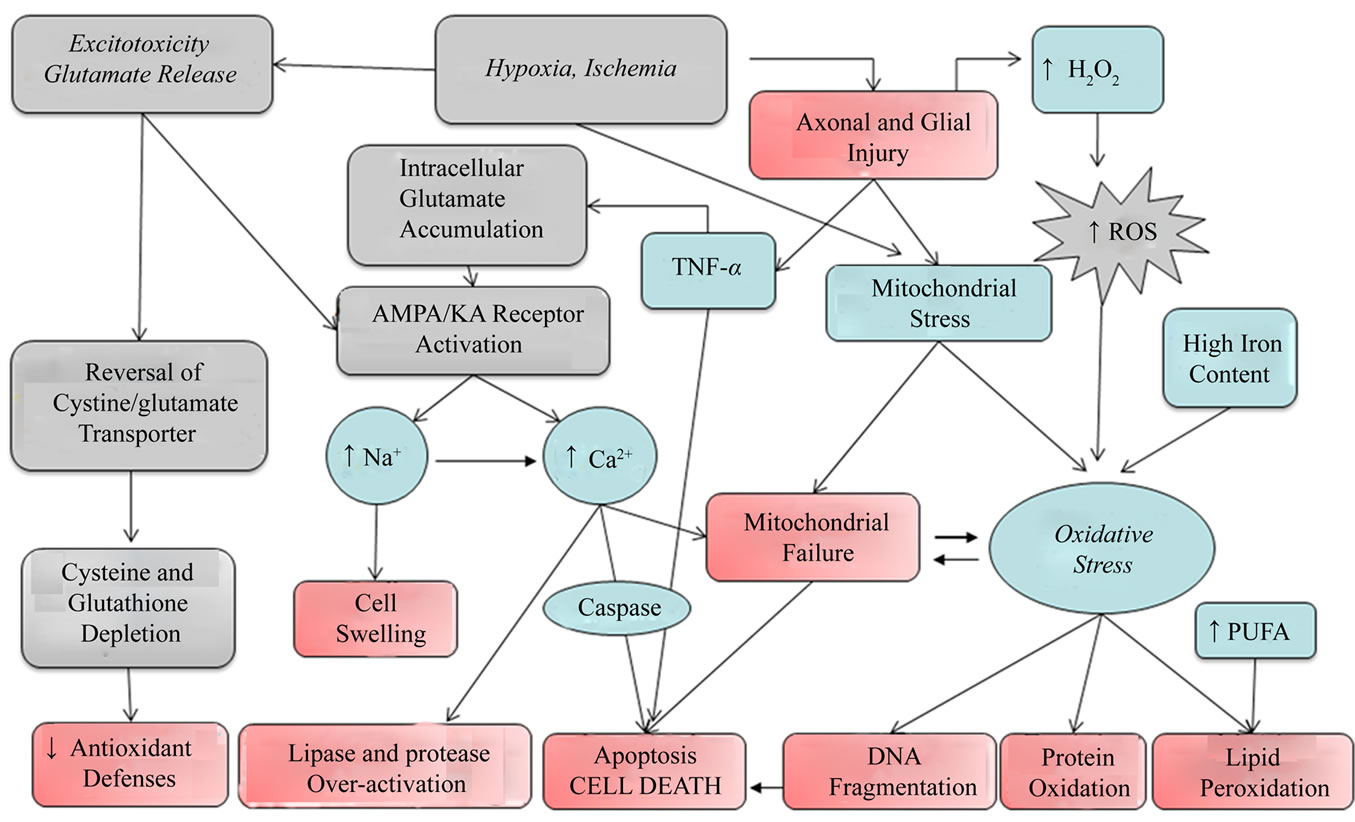
Figure 1. Oligodendrocyte injury mechanisms. Schematic representation portrays different mechanisms of injury to oligodendrocytes. Hypoxia/ischemia, excitotoxicity, oxidative stress and cytokines are considered to be important players exerting cytotoxic effects to the oligodendrocytes. Glutamate mediated excitotoxicity occurs directly through activation of AMDA and KA receptors, and indirectly by causing intracellular depletion of cysteine and glutathione, thereby making them more prone to oxidative injury. Hypoxia/ischemia causes a direct injury to axons and glia causing TNF-α release (inflammatory cytokine) and indirectly increases excitotoxicity; increases mitochondrial stress. TNF-α causes apoptosis and necrosis in oligodendrocyte by caspase related pathways and indirectly by increasing intracellular glutamate concentration by interrupting astrocytes uptake. H2O2 produced in higher levels from injured microglia increases ROS production which proceeds to cause oxidative stress and aftermath features like DNA fragmentation, protein oxidation, lipid peroxidation, apoptosis. High iron stores and high level of PUFA in OLGs make them prone to oxidative stress and lipid peroxidation respectively.
can also cause glial death indirectly by inducing the release of toxic agents like TNF-α from microglia [14].
3.3. Inflammatory Cytokines
Cytokines, a family of proteins related to immune system, are known to cause oligodendrocyte injury accompanying autoimmunological pathology; studies have mainly focused on TNF-α, IFN-γ and Interleukin-1β (IL-β). TNF-α produced by immune system cells and by neurons, astrocytes and microglia. It induces both apoptotic and necrotic death of OLGs and inhibition of caspase-1 and caspase-3 protects them against TNF-α-induced apoptosis [11]. Mutations or breakages in mitochondrial DNA, damage to oligodendrocyte mitochondria or inhibition of oxidative metabolism could also deplete the supply of energy for biosynthesis of the components of mature membrane sheets and/or trigger oligodendroglial degeneration [45]. TNF-α regulates glutamate transport capacity in astrocytes, which leads to the accumulation of glutamate, increasing excitotoxic damage to OLGs and neuron [46]. TNF-α pro-apoptotic actions on OLGs are attributed to decreased IGF-1 expression and activity which can be rescued for external application of IGF-1 both in vivo and in vitro [11]. TNF-α may directly damage OLGs by forming peroxides of the long-chain fatty acids in lipids or via generation of ceramide which is cytotoxic to OLGs [47]. IFN-γ produced by lymphocytes in MS lesions plays a role in apoptosis of oligodendrcoytes both in vivo and in vitro. TNF-α increases the IFN-γ induced death of OPCs and caspase inhibition partially suppresses it [11]. IL-1β, a prominent microglial pro-inflammatory cytokine adversely affects oligodendrocyte survival. It is a potent regulator of the production of various matrix metalloproteinase (MMP) members which are implicated in demyelination and axonal loss in CNS diseases [48].
4. Oligoprotectants and White Matter Protecting Agents
Research on oligoprotectants has been going on for a long time, since oligopathy has long been noted in demyelinating diseases like multiple sclerosis, periventricular leukomalacia (PVL), acute disseminated encephalomyelitis (ADEM). Later on, with the focus on protecting neurovascular unit in stroke and other neurological diseases more researches have been carried out aimed at protecting oligodendrocytes. Hereby, a few drugs and factors that have proved useful in protecting OLGs and white matter as a whole using different models are discussed, a summary of which can be seen in Table 1.
4.1. Antibacterial Agent—Minocycline
Minocycline, a second-generation tetracycline, has shown beneficial effects in a wide range of acute neurological injuries. In rodent brain ischemic models, it showed anti-inflammatory effects by inhibiting immune mediators such as microglia. It has shown to attenuate hypoxia/ ischemia-induced white matter injury in the neonatal rat though report of directly protecting OLGs against ischemic stress in adult rodent stroke model is missing. It has also been seen that delayed minocycline treatment reduced long-term functional deficits as well as white matter injury in ET-1-induced rat ischemia model. Despite reports from animal models clinical trial using mino cycline in Amyotrophic lateral sclerosis (ALS) patients, failed to show efficacy, this could be attributed to the long term use of minocycline, a powerful metalloproteinase inhibitor. It has been suggested that long-term suppression of metalloproteinases may be detrimental for neurovascular homeostasis [49].
4.2. Antioxidants and Free Radical Scavengers
Antioxidants and free radical scavengers have proved beneficial to OLGs which show a great vulnerability to oxidative stress. Body’s endogenous component and various synthetic drugs have been tried for use in preventing damage to OLGs and a few of them which proved beneficial have been listed below.
4.2.1. Endogenous Component-CoenzymeQ10 (CoQ10)
CoQ10 an endogenous component of the mitochondrial electron transport chain is known to attenuate the peroxidation of lipids and protect neurons against free radical in both in vitro primary cell culture and experi mental animals. It was shown that it can protect OLGs against TNF-α using primary culture of rat glial cells. In the same study, CoQ10 restored the numbers of mature myelin basic protein-positive cells and the ability of the OLGs to form membrane sheets [47]. However, there is published case of an in vivo model of focal and global ischemia in rats where CoQ10 failed to protect brain [50].
4.2.2. Synthetic Drugs
Various synthetic drugs in the market possessing antioxidant activity are being screened for their effectiveness in protecting the OLGs in different injury models. Here, we have listed some drugs that have proved to be useful in the researches aimed at protecting OLGs and white matter injury.
4.2.2.1. Ebselen
Ebselen is a synthetic seleno-organic drug with glutathione peroxidase like activity [51]. Imai et al. using rat transient ischemia models showed that ebselen reduced axonal damage and oligodendrocyte pathology [24].
4.2.2.2. Edaravone
Edaravone, a radical scavenger of hydroxyl radicals, has


Table 1. Oligoprotectants and white matter protecting agents.
shown to decrease the loss of OLGs and attenuate white matter lesions through endothelial protection and free radical scavenging effects in a chronic hypoperfusion model in rats. Randomized clinical trial setting has also shown significant functional improvement in acute ischemic stroke patients [52].
4.2.2.3. N-acetyl cysteine (NAC)
NAC is a free radical scavenger and an antioxidant that can modulate gene transcription and apoptosis. Cammer using in vitro primary rat cell culture showed that NAC was able to restore the numbers of mature myelin basic protein-positive cells and the ability of the OLGs to form membrane sheets following studies that it protected neuroblastoma cultures against oxidative stress and cytotoxicity [47].
4.2.2.4. Methylprednisolone
Methylprednisolone, an anti-inflammatory and antioxidant drug, is used to treat a variety of neurological disorders involving white matter injury like MS, acute disseminated encephalomyelitis, and SCI. It selectively inhibits oligodendrocyte but not neuronal cell death via a receptor-mediated action and may be a mechanism for its limited protective effect after SCI. It was shown to attenuate oligodendrocyte death in a dose-dependent manner in staurosporine or AMPA model of injury through glucocorticoid receptor dependent mechanism; however, same dose failed to prevent the neurons [53].
4.2.2.5. Phenyl-N-tert-butyl-nitrone (PBN)
PBN a free radical scavenger has proved to be useful in preventing oligodendrocyte damage in various models. Using rat MCAO model, Irving et al. demonstrated that a free radical scavenger PBN reduced injury to the OLGs through the reduced number of tau-positive OLGs in the sub-cortical white matter of the ischemic hemisphere in PBN treated rats. Lin et al. using hypoxia-ischemia model in the neonatal rat brain displayed PBN treatment protected both OLGs and axons from ischemic insults. In the same model, it was seen that PBN also inhibits upregulation of inflammatory cytokines such as IL-1β, TNF-α and inducible NOS mRNA expression [60].
4.2.2.6. Ramipril
Ramipril, an angiotensin converting enzyme inhibitor, has shown to attenuate white matter lesions against chronic hypoperfusion ischemia owing to its free radical scavenging effect in rat models [62].
4.3. Cord Blood
It has been shown that infusion of human umbilical cord blood cells protects striatal white matter tracts in vivo and directly protects mature primary oligodendrocyte cultures from oxygen glucose deprivation (OGD) [55].
4.4. Endogenous Factors
Different endogenous factors secreted in animals have positive influences in OLGs. The PDGF, basic-fibroblast growth factor (b-FGF), and IGF-1 are known to influence oligodendrocyte lineage cell proliferation and survival [64]. A few of these endogenous factors have shown to afford protection to the OLGs against different stress conditions. The promising results from endogenous factors have been listed below.
4.4.1. Ciliary Neurotrophic Factor (CNTF)
CNTF, a trophic factor found in astrocytes, has shown to inhibit TNF-α apoptotic death of oligodendrocytes in an in vitro model. It plays a role in survival of OLGs during development and may help in developing treatment strategies aimed at demyelinating diseases [54].
4.4.2. Insulin-Like Growth Factor-1 (IGF-1)
IGF-1 is known for its neuroprotective effects and is taken as a promising candidate for neuromuscular diseases [65]. It plays an immense role in OLGs right from the developmental stage, enhanching OPC survival to maturation and stimulating myelination [64,66,67]. IGF- 1 also known as the survival factor for OLGs [68], when applied externally has shown to decrease the pro-apo ptotic actions of TNF-α [11].
4.4.3. Interleukin-11 (IL-11)
IL-11, an astrocyte-derived which is localized in the active and silent lesions of MS, is linked with increase in OLG number and remyelination in the early stages. It potentiates oligodendrocyte survival and maturation, and myelin formation. In vitro human cell cultures have shown that immature OLGs express IL-11Rα receptors and IL-11 has been linked with enhanced oligodendrocyte survival and maturation. Rodent CNS co-culture studies have linked it to increased myelination, thus implicating IL-11 in oligodendrocyte viability, maturation, and myelination [15].
4.4.4. Nerve Growth Factor (NGF)
NGF has long been known for causing neurite outgrowth and hence the name nerve growth factor. It protects OLGs from TNF-α mediated cytotoxicity. Nerve growth factor also prevented the tumor necrosis factor-induced loss of mitochondrial membrane potential [61].
4.5. Glutamate Receptor Antagonist
Glutamate excitotoxicity related death of oligodendrocyte is seen in many disease states and thus exploiting the receptor antagonists have proved to be beneficial in different experiments. The antagonists of AMPA/KA and NMDA receptor types associated with glutamate signaling have shown significant reduction of OLG death in hypoxia/ischemia models. Hence, they may prove beneficial targets for prevention of ischemic stroke and related OLG deaths.
4.5.1. AMPA/KA Antagonist—NBQX
The AMPA/kainate receptor mechanism has recently been implicated in white matter injury following white matter ischemia. NBQX, an AMPA/kainate receptor antagonist, has shown to reduce ischemia-induced white matter injury and improve locomotor function in a rat ischemic spinal cord model [24].
4.5.2. NMDA Antagonist—Memantine
Memantine, an uncompetitive NMDA receptor antagonist, is a licensed drug for moderate-to-severe Alzheimer’s disease in US and EU. Memantine attenuated white matter injury in a rat model of periventricular leukomalacia and may also be protective to white matter in ischemia. It has been reported that memantine reduced ischemic damage to mature and precursor OLGs in brain slices following assessment by patch-clamp system [60].
4.6. Hormones
Even the hormones like erythropoietin and melatonin have proved beneficial in improving recovery of injured white matter through increased oligodendrogenesis and through reduction of oxidative stress. Also thyroxine is considered to be vital for survival of OLGs and it is known to trigger the maturation process of OLGs [69].
4.6.1. Erythropoietin
The hormone erythropoietin made by the kidney cells under hypoxic conditions acts on stem cells in the bone marrow to increase the production of red blood cells. Erythropoietin mediated neuroprotection against neonatal hypoxic-ischemic brain injury has been known since 2003. It promotes differentiation and maturation of OLGs and is also expected to affect the ability of OLGs to promote myelin repair in normal and damaged CNS [70]. Increased oligodendrogenesis, recovery of injured white matter and improved neurological function recovery by delayed and repeated administration of erythropoietin was seen after neonatal hypoxic-ischemic brain injury in rats although the delayed administration of erythropoietin did not decrease infarct volume [56].
4.6.2. Melatonin
Melatonin, a naturally occurring hormone, has been in use for the treating sleep related disorders. It has been found to reduce oxidative damage in neurons and OLGs through antioxidant and radical scavenging activity as seen in an experiment using mouse middle cerebral artery occlusion (MCAO) model using amyloid precursor protein (APP) and microtubule associated protein Tau-1 as markers for evaluating post-ischemic disrupted axonal flow and oligodendrocyte pathology respectively through immunohistochemical studies. It is known to reduce infarct volumes and enhance neurobehavioral and electrophysiological recoveries [59].
4.7. HSP90 Inhibitors
Heat shock protein 90 (HSP90) present in OPCs surface has been known to form autoantibodies in Multiple Sclerosis. HSP90 inhibitors radicicol and 17-allylamino- 17-demethoxygeldanamycin (17-AAG) at non-cytotoxic doses target cell-surface HSP90 in OPCs and through competitive inhibition of receptors prevent HSP90-antibody-induced OPC death thus protecting them against antibody attack. They also have shown protective effect on adult OLGs [57].
4.8. Immunomodulator (Synthetic)-Linomide
Bao et al. using primary cultures of rat glial cells showed that a synthetic immunomodulator linomide prevents OLGs damage by suppressing nitric oxide production in microglia and astrocytes [58].
4.9. Sodium Channel Blockers
Ischemic injury in the gray matter is associated with excitatory amino acid neurotransmitters release, and in the white matter is associated with intracellular sodium accumulation. The effect of sodium channel blocking drugs phenytoin, carbamazepine and diazepam on anoxia-induced injury in CNS white matter in vitro rat optic nerve preparation has shown to offer protection against anoxic injury at concentrations much lower than that required for treating epilepsy [63]. Sodium channel blockers represent an attractive therapeutic target because of their ability to simultaneously interfere indirectly with several calcium sourcing pathways [71].
5. Conclusions
Oligodendrocytes are most vulnerable cells of the brain with highest energy requirement, high iron stores, and low glutathione content; implicated primarily in demyelinating diseases and secondarily in neuropathies. The maintenance of OLGs at functional level is necessary for proper functioning of neuronal pathways, the loss of which may lead to multiple functional loss, axonal degeneration and/or neuronal death. Therefore, the neurocentric view of neuroprotection has widened to embrace the glial cells. In order to get along with neuroprotection, which has been the central focus for long, much work needs to be done on finding ways of protecting the glial cells. For this, a better understanding of the mechanisms by which glial cells death ensues in various disease states is utmost important. A better understanding of the injury mechanisms of OLGs should be beneficial as we move along the path of protecting the long shadowed glial cells hand in hand with neuroprotection as the concept of the neurogliovascular unit flourishes.
REFERENCES
- K. Arai and E. H. Lo, “Oligovascular Signaling in White Matter Stroke,” Biological & Pharmaceutical Bulletin, Vol. 32, No. 10, 2009, pp. 1639-1644. doi:10.1248/bpb.32.1639
- M. Bastide, T. Ouk, F. Plaisier, O. Pétrault, S. Stolc and R. Bordet, “Neurogliovascular unit after cerebral ischemia: is the vascular wall a pharmacological target,” Psychoneuroendocrinology, Vol. 32, Supplement 1, No. 1, 2007, pp. S36-S39.
- B.V. Zlokovic, “Neurodegeneration and the neurovascular unit,” Nature Medicine, Vol. 16, No.12, 2010, pp. 1370-1371. doi:10.1038/nm1210-1370
- N. Baumann and D. Pham-Dinh, “Biology of Oligoden-drocyte and Myelin in the Mammalian Central Nervous System,” Physiological Reviews, Vol. 81, No. 2, 2001, pp. 871-927.
- G. Bartzokis, “Age-related myelin breakdown: a develop-mental model of cognitive decline and Alzheimer’s disease,” Neurobiology of Aging, Vol. 25, No. 1, 2004, pp. 5-18. doi:10.1016/j.neurobiolaging.2003.03.001
- J. van Horssen, M. E. Witte, G. Schreibelt and H. E. de Vries, “Radical changes in multiple sclerosis pathogenesis,” Biochimica et Biophysica Acta, Vol. 1812, No. 2, 2010, pp. 141-150.
- M. Bradl and H. Lassmann, “Oligodendrocytes: biology and pathology,” Acta Neuropathologica, Vol. 119, No. 1, 2010, pp. 37-53. doi:10.1007/s00401-009-0601-5
- C. Richter-Landsberg and U. Vollgraf, “Mode of cell injury and death after hydrogen peroxide exposure in cultured oligodendroglia cells,” Experimental Cell Research, Vol. 244, No. 1, 1998, pp. 218-229. doi:10.1006/excr.1998.4188
- M. Nedergaard, J. J. Rodríguez and A. Verkhratsky, “Glial calcium and diseases of the nervous system,” Cell Calcium, Vol. 47, No. 2, 2010, pp. 140-149. doi:10.1016/j.ceca.2009.11.010
- D. Zieve, “Multiple Aclerosis,” Pubmed Health, 2010. http://www.ncbi.nlm.nih.gov/pubmedhealth/PMH0001747/
- C. A. Tegla, C. Cudrici, V. Rus, T. Ito, S. Vlaicu, A. Singh and H. Rus, “Neuroprotective effects of the complement terminal pathway during demyelination: implica-tions for oligodendrocyte survival,” Journal of Neuroimmunology, Vol. 213, No. 1-2, 2009, pp. 3-11. doi:10.1016/j.jneuroim.2009.06.006
- A. Jana and K. Pahan, “Oxidative Stress Kills Human Primary Oligodendrocytes via Neutral Sphingomyelinase: Implications for Multiple Sclerosis,” Journal of Neuroimmune Pharmacology, Vol. 2, No. 1, 2007, pp. 184-193. doi:10.1007/s11481-007-9066-2
- M. Vanmeeteren, J. Hendriks, C. Dijkstra and E. Vantol, “Dietary compounds prevent oxidative damage and nitric oxide production by cells involved in demyelinating disease,” Biochemical Pharmacology, Vol. 67, No. 5, 2004, pp. 967-975. doi:10.1016/j.bcp.2003.10.018
- C. Matute, E. Alberdi, M. Domercq, M.-V. SánchezGómez, A. Pérez-Samartín, A. Rodríguez-Antigüedad, and F. Pérez-Cerdá, “Excitotoxic damage to white matter,” Journal of Anatomy, Vol. 210, No. 6, 2007, pp. 693-702. doi:10.1111/j.1469-7580.2007.00733.x
- Y. Zhang, C. Taveggia, C. Melendez-Vasquez, S. Einheber, C. S. Raine, J. L. Salzer, C. F. Brosnan and G. R. John, “Interleukin-11 potentiates oligodendrocyte survival and maturation, and myelin formation,” The Journal of Neuroscience, Vol. 26, No. 47, 2006, pp. 12174- 12185. doi:10.1523/JNEUROSCI.2289-06.2006
- S. A Back, N. L. Luo, N. S. Borenstein, J. M. Levine, J. J. Volpe and H. C. Kinney, “Late oligodendrocyte progenitors coincide with the developmental window of vulnerability for human perinatal white matter injury,” The Journal of Neuroscience, Vol. 21, No. 4, 2001, pp. 1302-1312.
- M. V. Johnston, A. Ishida, W. N. Ishida, H. B. Matsushita, A. Nishimura and M. Tsuji, “Plasticity and injury in the developing brain,” Brain & Development, Vol. 31, No. 1, 2009, pp. 1-10. doi:10.1016/j.braindev.2008.03.014
- Y. Pang, L. Campbell, B. Zheng, L. Fan, Z. Cai and P. Rhodes, “Lipopolysaccharide-activated microglia induce death of oligodendrocyte progenitor cells and impede their development,” Neuroscience, Vol. 166, No. 2, 2010, pp. 464-475. doi:10.1016/j.neuroscience.2009.12.040
- K. G. Lee, “Periventricular Leukomalacia,” Pubmed Health, 2009. http://www.ncbi.nlm.nih.gov/pubmedhealth/PMH0004493/
- J. J. Volpe, H. C. Kinney, E. J. Frances,and P. A. Rosenberg, “The developing oligodendrocyte: key cellular target in brain injury in the premature infant,” International Journal of Developmental Neuroscience, Vol. 29, No. 4, 2011, pp. 423-440.
- D. B. Hoch, “Stroke,” Pubmed Health, 2010. http://www.ncbi.nlm.nih.gov/pubmedhealth/PMH0001740/
- A. S. Hazell, “Excitotoxic mechanisms in stroke: an Up-date of concepts and treatment strategies,” Neurochemistry International, Vol. 50, No. 7-8, 2007, pp. 941-953. doi:10.1016/j.neuint.2007.04.026
- W. Pan and A. J. Kastin, “Tumor necrosis factor and stroke: role of the blood-brain barrier,” Progress in Neurobiology, Vol. 83, No. 6, 2007, pp. 363-374. doi:10.1016/j.pneurobio.2007.07.008
- D. Dewar, S. M. Underhill and M. P. Goldberg, “Oligodendrocytes and Ischemic Brain Injury,” Journal of Cerebral Blood Flow & Metabolism, Vol. 23, No. 1, 2003, pp. 263-274. doi:10.1097/00004647-200303000-00001
- E. H. Lo, T. Dalkara and M. A. Moskowitz, “Mechanisms, challenges and opportunities in stroke,” Nature Reviews Neuroscience, Vol. 4, No. 5, 2003, pp. 399-415. doi:10.1038/nrn1106
- D. Zieve, “Spinal cord trauma,” Pubmed Health, 2010. http://www.ncbi.nlm.nih.gov/pubmedhealth/PMH0002061/
- R. Talac, J. A. Friedman, M. J. Moore, L. Lu, E. Jabbari, A. J. Windebank, B. L. Currier and M. J. Yaszemski, “Animal models of spinal cord injury for evaluation of tissue engineering treatment strategies,” Biomaterials, Vol. 25, No. 9, 2004, pp. 1505-1510. doi:10.1016/S0142-9612(03)00497-6
- G. D. Carlson and C. Gorden, “Current developments in spinal cord injury research,” The Spine Journal, Vol. 2, No. 1, 2002, pp. 116-128. doi:10.1016/S1529-9430(01)00029-8
- Y. H. Ahn, G. Lee and S. K. Kang, “Molecular insights of the injured lesions of rat spinal cords: Inflammation, apoptosis, and cell survival,” Biochemical and Biophysical Research Communications, Vol. 348, No. 1, 2006, pp. 560-570. doi:10.1016/j.bbrc.2006.07.105
- “Leukodystrophies,” Medline Plus, 2011. http://www.nlm.nih.gov/medlineplus/leukodystrophies.html.
- R. Káradóttir and D. Attwell, “Neurotransmitter receptors in the life and death of oligodendrocytes,” Neuroscience, Vol. 145, No. 4-5, 2007, pp. 1426 -1438.
- B. D. Butts, C. Houde and H. Mehmet, “Maturation-Dependent sensitivity of oligodendrocyte lineage cells to apoptosis: implications for normal development and disease,” Cell Death and Differentiation, Vol. 15, No. 1, 2008, pp. 1178-1186. doi:10.1038/cdd.2008.70
- A. D. Roth, G. Ramírez, R. Alarcón and R. V. Bernhardi, “Oligodendrocytes damage in Alzheimer’s disease : Beta amyloid toxicity and inflammation,” Biological Research, Vol. 38, No. 1, 2005, pp. 381-387.
- D. B. Merrill, “Schizophrenia,” Pubmed Health, 2010. http://www.ncbi.nlm.nih.gov/pubmedhealth/PMH0001925/
- N. A. Uranova, V. M. Vostrikov, O. V. Vikhreva, I. S. Zimina, N. S. Kolomeets and D. D. Orlovskaya, “The role of oligodendrocyte pathology in schizophrenia,” The International Journal of Neuropsychopharmacology, Vol. 10, No. 4, 2007, pp. 537-545. doi:10.1017/S1461145707007626
- P. R. Hof, V. Haroutunian, C. Copland, K. L. Davis and J. D. Buxbaum, “Molecular and cellular evidence for an oligodendrocyte abnormality in schizophrenia,” Neurochemical Research, Vol. 27, No. 10, 2002, pp. 1193-1200. doi:10.1023/A:1020981510759
- D. Segal, J. R. Koschnick, L. H. A. Slegers and P. R. Hof, “Oligodendrocyte pathophysiology: a new view of schizophrenia,” The International Journal of Neuropsychopharmacology, Vol. 10, No. 4, 2007, pp. 503-511. doi:10.1017/S146114570600722X
- A. Verkhratsky and V. Parpura, “Recent advances in (patho) physiology of astroglia,” Acta Pharmacologica Sinica, Vol. 31, No. 1, 2010, pp. 1044-1054. doi:10.1038/aps.2010.108
- C. Mihai and B. Jubelt, “Post-infectious encephalomyelitis,” Current Neurology and Neuroscience Reports, Vol. 5, No. 1, 2005, pp. 440-445. doi:10.1007/s11910-005-0031-2
- K. Sano, K. Morii, M. Sato, H. Mori and R. Tanaka, “Radiation-induced diffuse brain injury in the neonatal rat model-radiation-induced apoptosis of oligodendrocytes,” Neurologia Medico-Chirurgica, Vol. 40, No. 10, 2000, pp. 495-499. doi:10.2176/nmc.40.495
- A. Ernst, A. Stolzing, G. Sandig and T. Grune, “Antioxidants effectively prevent oxidation-induced protein damage in OLN 93 cells,” Archives of Biochemistry and Biophysics, Vol. 421, No. 1, 2004, pp. 54-60. doi:10.1016/j.abb.2003.10.008
- H. Mann, M. T. McCoy, J. Subramaniam, H. V. Remmen, and J. L. Cadet, “Overexpression of superoxide dismutase and catalase in immortalized neural cells: toxic effects of hydrogen peroxide,” Brain Research, Vol. 770, No. 1, 1997, pp. 163-168. doi:10.1016/S0006-8993(97)00768-3
- C. Matute, “Oligodendrocyte NMDA receptors: a novel therapeutic target,” Trends in Molecular Medicine, Vol. 12, No. 7, 2006, pp. 289-292. doi:10.1016/j.molmed.2006.05.004
- E. A. Leuchtmann, A. E. Ratner, R. Vijitruth, Y. Qu and J. W. Mcdonald, “AMPA receptors are the major mediators of excitotoxic death in mature oligodendrocytes,” Neurobiology of Disease, Vol. 14, No. 1, 2003, pp. 336-348. doi:10.1016/j.nbd.2003.07.004
- W. Cammer and H. Zhang, “Maturation of oligodendrocytes is more sensitive to TNF-α than is survival of precursors and immature oligodendrocytes,” Journal of Neuroimmunology, Vol. 97, No. 1, 1999, pp. 37-42. doi:10.1016/S0165-5728(99)00045-4
- W. Chadwick, T. Magnus, B. Martin, A. Keselman, M. P. Mattson and S. Maudsley, “Targeting TNF-α receptors for neurotherapeutics,” Trends in Neurosciences, Vol. 31, No. 10, 2008, pp. 504-511. doi:10.1016/j.tins.2008.07.005
- W. Cammer, “Protection of cultured oligodendrocytes against tumor necrosis factor-α by the antioxidants coenzyme Q10 and N-acetyl cysteine,” Brain Research Bulletin, Vol. 58, No. 6, 2002, pp. 587-592. doi:10.1016/S0361-9230(02)00830-4
- J. L. Takahashi, F. Giuliani, C. Power, Y. Imai and V. W. Yong, “Interleukin-1β promotes oligodendrocyte death through glutamate excitotoxicity,” Annals of Neurology, Vol. 53, No. 5, 2003, pp. 588-595. doi:10.1002/ana.10519
- T. Y. Yune, J. Y. Lee, G. Y. Jung, S. J. Kim, M. H. Jiang, Y. C. Kim, Y. J. Oh, G. J. Markelonis and T. H. Oh, “Minocycline alleviates death of oligodendrocytes by inhibiting pro-nerve growth factor production in microglia after spinal cord injury,” The Journal of Neuroscience, Vol. 27, No. 29, 2007, pp. 7751-7761. doi:10.1523/JNEUROSCI.1661-07.2007
- H. Li, G. Klein, P. Sun and A. M. Buchan, “CoQ10 fails to protect brain against focal and global ischemia in rats,” Brain Research, Vol. 877, No. 1, 2000, pp. 7-11. doi:10.1016/S0006-8993(00)02609-3
- Y. Nakamura, Q. Feng, T. Kumagai, K. Torikai, H. Ohigashi, T. Osawa, N. Noguchi, E. Niki and K. Uchida, “Ebselen, a Glutathione Peroxidase Mimetic Seleno-Organic Compound, as a Multifunctional Antioxidant,” The Journal of Biological Chemistry, Vol. 277, No. 4, 2002, pp. 2687-2694. doi:10.1074/jbc.M109641200
- Y. Ueno, N. Zhang, N. Miyamoto, R. Tanaka, N. Hattori and T. Urabe, “Edaravone attenuates white matter lesions through endothelial protection in a rat chronic hypoperfusion model,” Neuroscience, Vol. 162, No. 2, 2009, pp. 317-327. doi:10.1016/j.neuroscience.2009.04.065
- J.-M. Lee, P. Yan, Q. Xiao, S. Chen, K.-Y. Lee, C. Y. Hsu and J. Xu, “Methylprednisolone protects oligodendrocytes but not neurons after spinal cord injury,” The Journal of Neuroscience, Vol. 28, No. 12, 2008, pp. 3141-3149. doi:10.1523/JNEUROSCI.5547-07.2008
- J. C. Louis, E. Magal, S. Takayama and S. Varon, “CNTF protection of oligodendrocytes against natural and tumor necrosis factor-induced death,” Science, Vol. 259, No. 5095, 1993, pp. 689-692. doi:10.1126/science.8430320
- D. D. Rowe, C. C. Leonardo, A. A. Hall, M. D. Shahaduzzaman, L. A. Collier, A. E. Willing and K. R. Pennypacker, “Cord blood administration induces oligodendrocyte survival through alterations in gene expression,” Brain Research, Vol. 1366, No. 1, 2010, pp. 172-188. doi:10.1016/j.brainres.2010.09.078
- S. Genc, K. Genc, A. Kumral and H. Ozkan, “White matter protection by erythropoietin: an emerging matter in the treatment of neonatal hypoxic-ischemic brain injury,” Stroke, Vol. 41, No. 11, 2010, p. e595. doi:10.1161/STROKEAHA.110.590844
- C. Cid and A. Alcazar, “Protection of oligodendrocyte precursor cells by low doses of HSP90 inhibitors in cell culture,” Experimental Neurology, Vol. 225, No. 7, 2010, pp. 29-33. doi:10.1016/j.expneurol.2009.11.017
- B. G. Xiao, X. F. Bail, G. X. Zhang, G. Hedlund and H. Link, “Linomide-mediated protection of oligodendrocytes is associated with inhibition of nitric oxide production and IL-1β expression in Lewis rat glial cells,” Neuroscience Letters, Vol. 249, No. 1, 1998, pp. 17-20. doi:10.1016/S0304-3940(98)00371-1
- E.-J. Lee, M.-Y. Lee, H.-Y. Chen, Y.-S. Hsu, T.-S. Wu, S.-T. Chen and G.-L. Chang, “Melatonin attenuates gray and white matter damage in a mouse model of transient focal cerebral ischemia,” Journal of Pineal Research, Vol. 38, No. 1, 2005, pp. 42-52. doi:10.1111/j.1600-079X.2004.00173.x
- [61] K. Arai and E. H. Lo, “Experimental models for analysis of oligodendrocyte pathophysiology in stroke,” Experimental & Translational Stroke Medicine, Vol. 1, No. 6, 2009.
- [62] R. Takano, S. Hisahara, K. Namikawa, H. Kiyama, H. Okano and M. Miura, “Nerve growth factor protects oli-godendrocytes from tumor necrosis factor-α-induced injury through Akt-mediated signaling mechanisms,” The Journal of Biological Chemistry, Vol. 275, No. 21, 2000, pp. 16360-16365. doi:10.1074/jbc.M910419199
- [63] J.-S. Kim, I. Yun, Y. B. Choi, K.-S. Lee and Y.-I. Kim, “Ramipril protects from free radical induced white matter damage in chronic hypoperfusion in the rat,” Journal of Clinical Neuroscience, Vol. 15, No. 2, 2008, pp. 174-178. doi:10.1016/j.jocn.2006.12.003
- [64] R. Fern, B. R. Ransom, P. K. Stys and S. G. Waxman, “Pharmacological protection of CNS white matter during anoxia: actions of phenytoin, carbamazepine and diazepam,” The Journal of Pharmacology and Experimental Therapeutics, Vol. 266, No. 3, 1993, pp. 1549-1555.
- [65] W. H. Lagarde, R. Benjamin, A. T. Heerens, P. Ye, R. I. Cohen, B. M. Moats-Staats and A. J. D’ercole, “A non-transformed oligodendrocyte precursor cell line, OL-1, facilitates studies of insulin-like growth factor-I signaling during oligodendrocyte development,” International Journal of Developmental Neuroscience, Vol. 25, No. 2, 2007, pp. 95-105. doi:10.1016/j.ijdevneu.2006.12.006
- [66] A. Musarò, G. Dobrowolny and N. Rosenthal, “The neuroprotective effects of a locally acting IGF-1 isoform,” Experimental Gerontology, Vol. 42, No. 1-2, 2007, pp. 76-80. doi:10.1016/j.exger.2006.05.004
- [67] P. Ye and A. J. D’Ercole, “Insulin-like growth factor I protects oligodendrocytes from tumor necrosis factor-α-induced injury,” Endocrinology, Vol. 140, No. 7, 1999, pp. 3063-3072. doi:10.1210/en.140.7.3063
- [68] F. A. McMorris, T. M. Smith, S. DeSalvo and R. W. Furlanetto, “Insulin-like growth factor I/somatomedin C: a potent inducer of oligodendrocyte development,” Proceedings of the National Academy of Sciences of the United States of America, Vol. 83, No. 3, 1986, pp. 822-826. doi:10.1073/pnas.83.3.822
- [69] J. L. Mason, S. Xuan, I. Dragatsis, A. Efstratiadis and J. E. Goldman, “Insulin-like growth factor (IGF) signaling through type 1 IGF receptor plays an important role in remyelination,” The Journal of Neuroscience, Vol. 23, No. 20, 2003, pp. 7710-7718.
- [70] S. A. Jones, D. M. Jolson, K. K. Cuta, C. N. Mariash and G. W. Anderson, “Triiodothyronine is a survival factor for developing oligodendrocytes,” Molecular and Cellular Endocrinology, Vol. 199, No. 1-2, 2003, pp. 49-60. doi:10.1016/S0303-7207(02)00296-4
- [71] M. Sugawa, Y. Sakurai, Y. Ishikawa-ieda, H. Suzuki and H. Asou, “Effects of erythropoietin on glial cell development; oligodendrocyte maturation and astrocyte proliferation,” Neuroscience Research, Vol. 44, No. 4, 2002, pp. 391-403. doi:10.1016/S0168-0102(02)00161-X
- [72] P. K. Stys, “White matter injury mechanisms,” Current Molecular Medicine, Vol. 4, No. 2, 2004, pp. 113-130. doi:10.2174/1566524043479220
NOTES
This study was supported by the National Natural Science Foundation of China (81070967/H0909); the Natural Science Foundation of Jiangsu province (No.BK2009296); the Initial Fund of China Pharmaceutical University (to Liao Hong).