Advances in Microbiology
Vol.4 No.12(2014), Article ID:49726,8 pages
DOI:10.4236/aim.2014.412086
Number of PCR Cycles and Magnesium Chloride Concentration Affect Detection of tet Genes Encoding Ribosomal Protection Proteins in Swine Manure
Gunilla Schmidt1, Jill Stiverson2, ystein Angen3, Zhongtang Yu2*
1The National Veterinary Institute, Technical University of Denmark, Copenhagen, Denmark
2Department of Animal Sciences, The Ohio State University, Columbus, Ohio, USA
3The Norwegian Veterinary Institute, Oslo, Norway
Email: *yu.226@osu.edu
Copyright © 2014 by authors and Scientific Research Publishing Inc.
This work is licensed under the Creative Commons Attribution International License (CC BY).
http://creativecommons.org/licenses/by/4.0/



Received 20 June 2014; revised 26 July 2014; accepted 19 August 2014
PCR is routinely used in detection of antibiotic resistance genes including different classes of tet and erm genes. It remains unknown how PCR conditions affect detection of resistance genes in terms of genetic diversity and prevalence. In this study, numbers of PCR cycles and MgCl2 concentrations were evaluated for their effect on the diversity and prevalence of the tet genes that encode ribosomal protection proteins (RPPs) in composted swine fecal samples using the degenerate Ribo2_new_FW/Ribo2-RV primer pair. Four MgCl2 concentrations and 3 cycle numbers were tested in a 4 × 3 factorial design. A clone library was constructed for each PCR condition combination, and randomly selected clones were sequenced to determine the genetic diversity and relative distribution of RPP tet genes. Significant differences in genetic diversity and prevalence of tet genes were found among the tested cycle numbers and MgCl2 concentration combinations. The results suggest that 35 PCR cycles and 7 mM MgCl2 allow for optimal detection of the tet genes in swine feces using the Ribo2_new_FW/Ribo2-RV primer pair and that this combination should be used for further assay optimization and validation. These results also suggest that PCR conditions should be taken into consideration when PCR conditions are chosen for ecological studies of tet genes and when the results are interpreted.
Keywords:Porcine, Antibiotic Resistance, Assay, Optimization, Reaction-Mix

Polymerase chain reaction (PCR) has been the choice of method to specifically and rapidly detect the presence and genetic diversity of genes of interest, including genes encoding antibiotic resistance in microbiome samples [1] [2] . Because it allows for detection of target genes carried by both culturable and nonculturable microbes, PCR can provide more accurate detection of the target genes than cultivation-based methods. PCR is a DNAbased technique, where specificity, sensitivity, and inclusiveness (inclusive amplification of all the intended target genes) are very important aspects. A number of factors, such as primers used, amount and quality of the template DNA, PCR thermal profile, and reaction mix, can affect the specificity, sensitivity, and inclusiveness. Most PCR assays are optimized for specific amplification of the target gene(s) by primarily testing different primer annealing temperatures and magnesium chloride (MgCl2) concentrations [3] . PCR additives, such as bovine serum albumin which attenuates amplification inhibition, or dimethyl sulfoxide (or glycerol, betaine, etc.) which reduce formation of secondary structures, are often added to the reaction mix to improve PCR efficiency or sensitivity. MgCl2 concentration plays an important role in PCR specificity, sensitivity and inclusiveness because the melting of double stranded DNA (including secondary structure formed from single stranded DNA) and annealing of the primers to the DNA template are affected by Mg2+. Furthermore, Taq DNA polymerase utilizes Mg2+ for activity [3] -[5] . It has been well established that too high a concentration of MgCl2 can increase the efficiency of the PCR amplification but may reduce the specificity, while too low a MgCl2 concentration can result in little or no PCR product [3] [4] . The optimal cycle number for specific PCR depends on the amount of template used, the primers used, and the efficiency of the PCR assay [5] . For a given amount of DNA template, if the cycle number is too low, then there will be little or no amplified products, but if it is too high non-specific products and high molecular weight smears may form [3] [5] .
When a single gene is amplified, both the PCR specificity and sensitivity are critical for amplification of the correct gene. However, when a class or family of related genes needs to be simultaneously detected using PCR and a pair of “universal” primers, inclusiveness also becomes important. In a previous study, it was noted that Fibrobacter succinogenes, a species of major cellulolytic bacteria in the rumen, could not be detected by PCR using a pair of universal bacterial primers that matches the 16S rRNA gene of that species perfectly [6] . In a recent study, we also repeatedly failed to detect Methanobacterium in samples collected from anaerobic digesters using a pair of Archaea domain-specific primers, although the primers matched the 16S rRNA gene sequences of this genus, and qPCR using genus-specific primers revealed high abundance of this genus [7] . Evidently, such biased PCR prevents actual diversity and relative abundance of target genes from being revealed. Classand group-specific primers have been commonly used in detecting the presence and diversity of antibiotic resistance genes, including tet genes and erm genes [8] -[13] . The tetracycline resistance tet genes either code for efflux proteins, ribosomal protection proteins (RPP), inactivating enzymes, or have unknown mechanisms [14] . There are currently 45 known classes of tetracycline resistance genes in addition to the recently described mosaic genes such as tet (O/W) and tet (W/O/W) [15] -[17] . The tetracycline resistance genes are often associated with other antibiotic resistance genes, especially erm genes that confer resistance towards macrolide, lincosamide, and streptogramin B antibiotics (MLSB) [18] . An updated overview of the tetracycline resistance genes can be found at http://faculty.washington.edu/marilynr/ [17] . MLSB antibiotics share overlapping binding sites and it is therefore common that resistance is conferred to more than one MLS drug. Macrolide resistance can be caused by rRNA methylases, rRNA methyltransferases, efflux systems, or antibiotic inactivation [18] . The rRNA methylase encoding erm genes confer resistance to MLSB antibiotics by inhibiting antibiotic binding to the ribosome [18] -[20] . There are currently 36 known methylase genes, one methyltransferasegene, 21 efflux genes, and 22 inactivating enzyme encoding genes (http://faculty.washington.edu/marilynr/ermwebA.pdf) [21] .
We hypothesized that PCR conditions, particularly numbers of PCR cycles and MgCl2 concentrations, can affect the inclusiveness of the PCR detection of individual antibiotic resistance genes, thus skewing characterization of the resistome actually present in a microbiome. To test this hypothesis, we evaluated the effect of numbers of PCR cycles and MgCl2 concentration on the detection of tet genes that encode ribosomal protection proteins (RPPs). The previously published degenerate primer pair Ribo2-FW/Ribo2-RV detects 7 classes of RPP tet genes, namely tet (B(P)), tet (M), tet (O), tet (Q), tet (S), tet (T), and tet (W), and they have successfully been used in detecting RPP tet genes in various samples [8] [22] -[24] . In the present study, the forward primer (Ribo2- FW) was altered so that the primer pair can detect the tet (32), tet (36), and tet (44) RPP genes which were discovered after the original Ribo2-FW/RV primers were designed [25] -[27] . Because this primer pair allows 10 different classes of RPP tet genes to be detected simultaneously, it can be useful in targeted metagenomic analysis of RPP tet genes. In the present study, the potential effects of different MgCl2 concentrations and PCR cycle numbers on diversity and proportional distribution of RPP tet genes were systematically examined using a factorial design. Our results showed that both of the parameters can have significant effect on the RPP tet genes that can be detected and their proportional distribution. The findings of the present study may aid in the design of future studies and interpretation of results, and can further be utilized for assay optimization and validation.
2.1. Samples
Swine manure samples were collected from the swine farm of Agricultural Technical Institute of The Ohio State University, which is a conventional research farm utilizing tetracyclines for growth promotion, disease prevention or therapy. The swine manure samples were composted as previously described and have been used in a previous study to investigate the successions of tet and erm genes during composting [28] . Briefly, a total of 3.3 kg swine manure was compost treated for 48 days. Fifty gram compost samples were collected on days 0, 17, and 48 and subsequently stored at −80˚C prior to further analysis. Metagenomic DNA was extracted using the repeated bead-beating plus column purification (RBB + C) method as described previously and the DNA integrity was confirmed by agarose (0.8%) gel electrophoresis [23] [28] [29] . In the present study, all the samples were pooled to create an “average” sample with diverse classes of tetracycline genes for PCR detection.
2.2. Primers
The primer sequences, targets, annealing temperatures, and amplicon lengths are given in Table1
Since the original primers Ribo2-FW and Ribo2-RV were published, three new classes of RPP tet genes, tet (32), tet (36) and tet (44), have been discovered. To expand the inclusiveness of the original Ribo2 primers, the sequences used to design the original Ribo2-FW and Ribo2-RV primers [8] were aligned with the tet (32), tet (36), and tet (44) sequences (respective accession numbers AJ295238, AJ514254, and FN594949) using ClustalX [30] . Then the new alignment of the RPP tet genes was aligned with the original Ribo2 primers. The Ribo2-RV matched the three new classes of RPP tet, but 2 of the 3 bases at the 3’ end of the Ribo2-FW primer did not match the tet (32), tet (36), and tet (44) sequences. Thus, the Ribo2-RW was modified at the 3’ end. The modified Ribo2-FW primer (referred to as Ribo2_new_FW) was evaluated for specificity using BLASTn and analyzed for formation of secondary structure and primer dimers using the Integrated DNA Technologies SciTools Oligoanalyzer (Integrated DNA Technologies, Inc., Coralville, IA). Both RPP tet primers, Ribo2_ new_FW and Ribo2-RV, were synthesized by Sigma-Aldrich (St. Louis, MO, United States).
2.3. PCR Amplification of RPP tet Genes
All PCR was performed using a 50 µl reaction volume with the same conditions except for varying MgCl2 concentrations and thermal cycle numbers. All the PCR contained the same amount (4.83 ng per reaction) of the same pooled DNA samples. Four MgCl2 concentrations (1.75 mM, 3 mM, 5 mM, and 7 mM) and three numbers of PCR cycles (20, 30 and 35) were tested in a 4 × 3 factorial design that resulted in 12 different combinations of PCR cycles and MgCl2 concentrations. Each PCR was run in duplicate along with a single non-template control with water replacing the DNA template. The following cycling conditions were used [23] : Initial denaturation for 4 minutes (min) at 94˚C, followed by 5 cycles of touchdown PCR consisting of 30 sec at 94˚C, 30 sec at 57˚C with 1˚C cycle−1 decrement, and 60 sec at 72˚C. Thereafter, there were 20, 30 or 35 cycles of PCR, each cycle consisting of 30 sec at 94˚C, 30 sec at 52˚C and 90 sec at 72˚C. There was a final elongation of 7 min at 72˚C.

Table 1. PCR primer sequences, targets, annealing temperatures, and amplicon length.
aPCR conditions are described in materials and methods.
2.4. Cloning and Sequencing
One clone library was constructed for each combination of cycle number and MgCl2 concentration that yielded the expected band (30 cycles × 5 mM MgCl2, 30 cycles × 7 mM MgCl2, 35 cycles × 3 mM MgCl2, 35 cycles × 5 mM MgCl2, and 35 cycles × 7 mM MgCl2). The amplicons were cloned using the TOPO TA cloning® kit for sequencing (Invitrogen, Life technologies, Grand Island, NY, United States) following the One Shot® Chemical Transformation Protocol. To maximize ligation efficiency, the 5-minute room temperature incubation step was extended to 15 minutes and 2 µl fresh PCR product was used. Clones were spread on Luria-Bertani (LB) plates containing 50 mg∙ml−1 ampicillin (Sigma-Aldrich, Saint Louis, MO, USA) and incubated for 24 hours at 37˚C. Sixty colonies from each clone library were randomly picked and inoculated into 5 ml LB broth with 50 mg∙ml−1 ampicillin and grown overnight at 37˚C. Positive clones were identified by growth in ampicillin based on the amp resistance gene on the pCR®4-TOPO® plasmid (Invitrogen, Life technologies, Grand Island, NY, United States).
Plasmid DNA extraction was performed using the QIAprep Miniprep kit (QIAGEN, Germantown, Maryland, United States) following the manufacturer’s instructions. Positive clones that contained the tet insert were confirmed by PCR using the M13 primers (Invitrogen, Life technologies, Grand Island, NY, United States). Sanger sequencing was performed on clones using DTCS Quick Start Kit (Beckman Coulter, Brea, CA, United States) on the Beckman GeneomeLab sequencer. A 5-minute heat treatment at 65˚C was preformed prior to the cycling sequencing reaction per Beckman protocol recommendations, and 5 mM M13f primer was used for sequencing. Base calling was manually verified and poor sequences were discarded. The ends of each sequence read with poor base calling were trimmed off using BioEdit (Ibis Biosciences, Carlsbad, CA, United States). The sequences compared to GenBank sequences using blastx to determine sequence identity to known tet genes in GenBank. The sequences that matched known tet genes were imported to Geneious version 5.6.4 (Geneious, Auckland, New Zealand) for alignment and tree construction. Trees were constructed using the Neighbor-joining method for each clone library. The sequence of ribosomal 5 s binding protein L5 of Thermus flavus (GBIS77826.1) was used as the outgroup to root the tree. The trees were exported in Newick format for use in UniFrac analysis [31] . Based on weighted UniFrac analysis, the UniFrac significance each pair of environments test was used to determine if the different PCR conditions (cycle numbers and MgCl2 concentrations) resulted in different detection (genetic diversity and proportion) of RPP tet genes.
No PCR amplicons were detected on agarose gels when only 20 cycles (excluding the five touchdown cycles) of PCR were performed at any of the four MgCl2 concentrations tested (data not shown). The abundance of RPP tet genes present in the pooled samples (containing both composted and uncomposted swine manure) might also be too low to produce a visible PCR band after 25 cycles of PCR amplification. The expected PCR amplicons were not found at 30 cycles with 1.75 mM or 3 mM MgCl2 and at 35 cycles with 1.75 mM MgCl2 (data not shown). These results suggest that 1.75 mM MgCl2 may be too low a concentration for reliable PCR detection of RPP tet genes in swine fecal samples using the Ribo2_new_FW/Ribo2-RV primers. The other combinations of PCR cycle numbers and MgCl2 concentrations (referred to as Cn/M combinations) yielded the expected PCR amplicons and the three new classes of RPP tet genes were also detected using the Ribo2_new_FW/Ribo2-RV primers (see Figure 1). The new primer Ribo2_new_FW may be used in future studies to allow detection of all known classes of RPP tet genes.
Different numbers of high-quality tet sequences were obtained from the different Cn/M combinations, ranging from 41 to 55 (see Figure 1). To compare the proportions of all the detected tet gene classes among the Cn/M combinations, the proportion of all the detected tet gene classes were normalized to a sum of 100% for each Cn/M combination. Differences in diversity (the classes detected) and relative abundance (proportions) were found among the different Cn/M combinations (see Figure 1 and Table 2). As hypothesized, the number of RPP tet gene classes detected increased with increasing PCR cycles and MgCl2 concentrations, with four, six, seven, eight and nine classes of RPP tet genes detected at 30/5, 30/7, 35/3, 35/5, and 35/7, respectively. The 35/5 combination yielded four DNA sequences that did not match tet genes. Interestingly, no non-specific sequences were found with the other Cn/M combinations including the least stringent 35/7 combination. This suggests that the increase in cycle number and MgCl2 concentration would necessarily increase the likelihood of non-specific
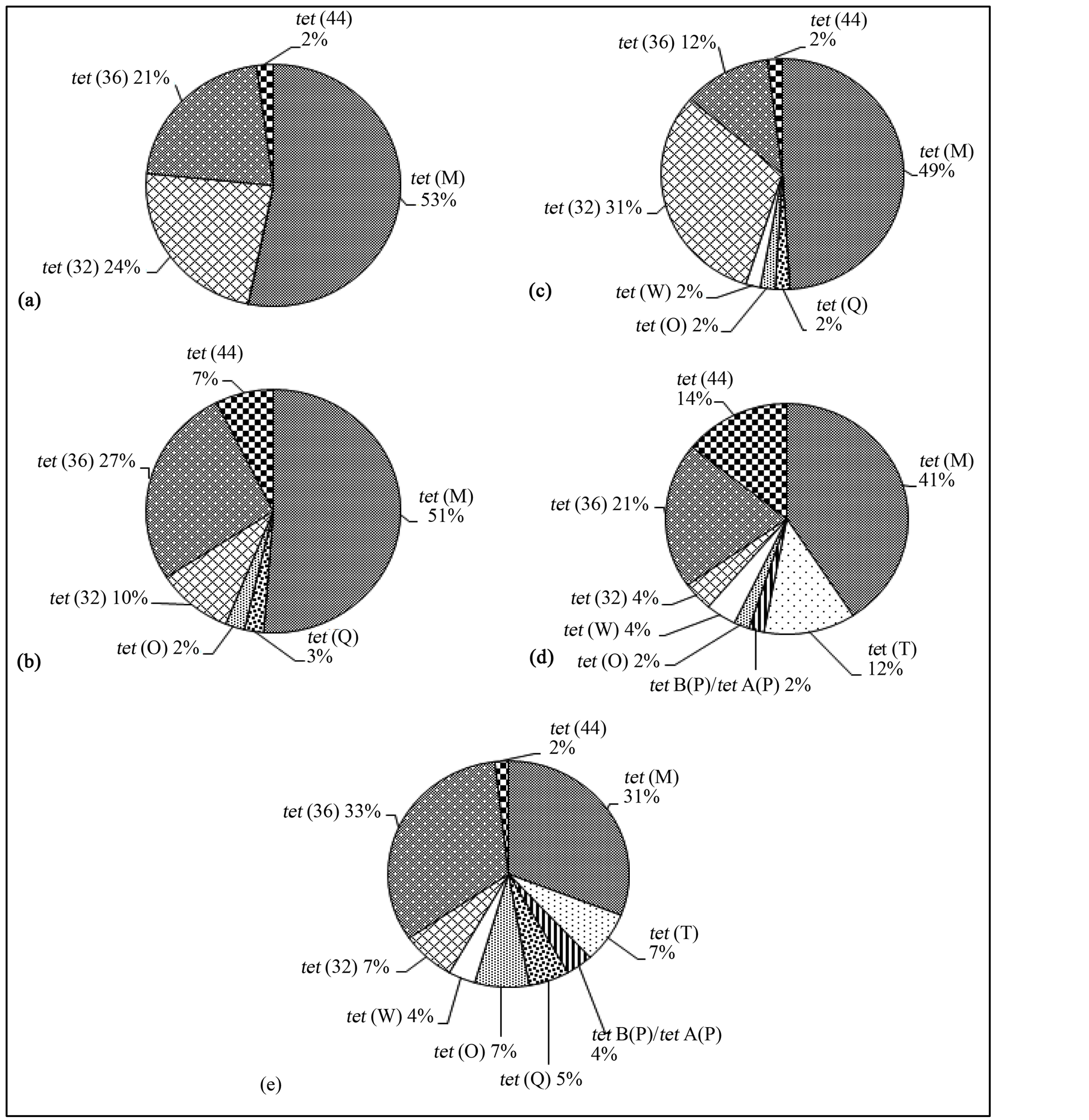
Figure 1. The classes of RPP tet detected in the swine manure and their proportion using different PCR cycle numbers and MgCl2 concentrations. (a) 30 cycles and 5 mM MgCl2 (n = 47); (b) 30 cycles and 7 mM MgCl2 (n = 41); (c) 35 cycles and 3 mM MgCl2 (n = 51); (d) 35 cycles and 5 mM MgCl2 (n = 49); (e) 35 cycles and 7 mM MgCl2 (n = 55). For comparison purpose, the proportions of the individual classes of RPP tet genes detected were normalized so that the total RRP tet gene summed to 100%.
Table 2. Matrix showing of the UniFrac significance test.
aCn/M: PCR cycle number/MgCl2 concentration (mM).
amplicons. UniFrac significance test revealed significant differences in the detection of RPP tet genes with respect to numbers of classes detected and their relative abundance among the Cn/M combinations (see Table 2).
The classes of tet genes detected and their proportions were affected to different degree by the different combinations of the PCR cycle numbers and the MgCl2 concentrations (see Figure 1). The tet (M), tet (32) and tet (36) genes were detected in all the PCR cycle number and MgCl2 combinations with tet (M) being the most predominant class followed by tet (36) and tet (32) in all the Cn/M combinations. The high tet (M) prevalence may be attributed to tet (M) being found in both Gram-positive and Gram-negative bacteria and having the broadest host range of all tet genes [23] . The tet (S) was not detected in the pooled swine manure samples by any of the Cn/M combinations, while tet (O) and tet (Q) became detectable at 30 cycles when MgCl2 concentration was increased from 5 to 7 mM. At 35 PCR cycles, the increase in MgCl2 concentration from 3 to 7 mM also resulted in detection of increased tet gene classes. At 5 mM MgCl2, simply increasing PCR cycle numbers from 30 to 35 enabled four additional classes of tet genes to be detected (W, O, B/P, and T). At 7 mM MgCl2, the increase in PCR cycle numbers from 30 to 35 also resulted in detection of three additional classes of tet genes that were otherwise not detected. Variations in cycle number and MgCl2 concentration also affected the proportion of the tet genes detected but to a different extent for different tet gene classes. For instance, the 2 mM increase (from 5 to 7 mM) in MgCl2 concentration at 30 cycles increased the proportion of tet (44) and tet (36) at the expense of tet (32). At 35 cycles, increase in MgCl2 concentration also affected the proportion of the tet gene classes detected but to different degrees for different tet gene classes. Such differential effect was also observed for the increase in PCR cycle number. For example, at 5 mM MgCl2 concentration, the increase in PCR cycles from 30 to 35 increased the proportion of tet (44) and tet (T) while decreasing that of tet (M). Differences in internal sequences among the tet gene classes may be one factor that contributes to difference in amplification efficiency and thus detection and proportion. In addition, a degenerate primer contains a pool of different primers. The proportion of a particular primer sequence within that pool can also affect primer annealing kinetics and subsequent amplification efficiency.
The original Ribo2-FW/RV primer pair was used in PCR analysis of swine manure that used 2 mM MgCl2 and 25 PCR cycles [8] . In that study, only three classes of RPP tet genes, tet (M), tet (O), and tet (W), were found. In another study using the original primer set, 1.75 mM MgCl2 and 5 touchdown PCR cycles followed by 30 cycles of regular PCR were used in qPCR to quantify total RPP tet genes [23] . Due to the alteration in the forward primer and the different sample types, the results from those studies cannot be directly compared to the results of the present study. Nevertheless, the current results indicate that together with touchdown cycles, increasing the cycle number to 35 and MgCl2 concentration to 7 mM may help detect the RPP tet genes present in samples without false negative results or compromising the specificity. Further optimization of this primer pair including establishment of the assay’s limit of detection should be conducted using the recommended cycle number and MgCl2 concentration for RPP tet gene detection in swine manure. In addition, although the effects of PCR cycle numbers and MgCl2 concentrations, among other factors, are conceivable, the results of the present study demonstrate the importance of conducting pilot studies investigating PCR assay parameters, such as cycle number and MgCl2 concentration, in order to establish the optimal conditions to accurately depict the resistome.
PCR bias has been well documented in amplification of 16S rRNA genes [32] . The results of this study also suggest that PCR bias also occurs with amplification of the individual classes of RPP tet genes. It should be noted that we did not evaluate PCR bias by using a pool of RPP tet genes with known proportions but we used the detection, or no-detection, of the RPP tet genes actually present in the samples as indicator of PCR bias. Such PCR bias should be taken into consideration when research results are interpreted within one study and when results are compared among different studies.
We would like to thank Amanda Gutek and Lingling Wang for their technical assistance. This research was supported in part by funding awarded to Z.Y. at the Ohio State University and the Strategic Research Council, Ministry for Food Agriculture and Fisheries, Denmark and the Faculty of Health and Medical Sciences, University of Copenhagen, Denmark.
No conflict of interest declared.
- Manuzon, M.Y., Hanna, S.E., Luo, H., Yu, Z., Harper, W.J. and Wang, H.H. (2007) Quantitative Assessment of the Tetracycline Resistance Gene Pool in Cheese Samples by Real-Time TaqMan PCR. Applied and Environmental Microbiology, 73, 1676-1677. http://dx.doi.org/10.1128/AEM.01994-06
- Alali, W.Q., Scott, H.M., Norby, B., Gebreyes, W. and Loneragan, G.H. (2009) Quantification of the blaCMY-2 in Feces from Beef Feedlot Cattle Administered Three Different Doses of Ceftiofur in a Longitudinal Controlled Field Trial. Foodborne Pathogens and Disease, 6, 917-924. http://dx.doi.org/10.1089/fpd.2009.0271
- Harris, S. and Jones, D.B. (1997) Optimisation of the Polymerase Chain Reaction. British Journal of Biomedical Science, 54, 166-173.
- Williams, F.J. (1989) Optimization Strategies for the Polymerase Chain Reaction. BioTechniques, 7, 762.
- Innis, M.A., Gelfand, D.H., Sninsky, J.J. and White, T.J. (1990) Optimization of PCRs. In: Innis, M.A., Gelfand, D.H., Sninsky, J.J. and White T.J., Eds., PCR Protocols: A Guide to Methods and Applications, Academic Press, London, 3-12.
- Larue, R., Yu, Z., Parisi, V.A., Egan, A.R. and Morrison, M. (2005) Novel Microbial Diversity Adherent to Plant Biomass in the Herbivore Gastrointestinal Tract, as Revealed by Ribosomal Intergenic Spacer Analysis and rrs gene Sequencing. Environmental Microbiology, 7, 530-543. http://dx.doi.org/10.1111/j.1462-2920.2005.00721.x
- Li, Y., Chen, P. and Yu, Z. (2014) Spatial and Temporal Variations of Microbial Community in a Mixed Plug-Flow Loop Reactor (MPFLR) Fed with Dairy Manure. Microbial Biotechnology, 7, 332-346. http://dx.doi.org/10.1111/1751-7915.12125
- Aminov, R.I., Garrigues, J.N. and Mackie, R.I. (2001) Molecular Ecology of Tetracycline Resistance: Development and Validation of Primers for Detection of Tetracycline Resistance Genes Encoding Ribosomal Protection Proteins. Applied and Environmental Microbiology, 67, 22-32. http://dx.doi.org/10.1128/AEM.67.1.22-32.2001
- Aminov, R.I., Chee-Sanford, J.C., Garrigues,J.N., Teferedegne, B., Krapac, I.J., White, B.A. and Mackie, R.I. (2002) Development, Validation, and Application of PCR Primers for Detection of Tetracycline Efflux Genes of Gram-Negative Bacteria. Applied and Environmental Microbiology, 68, 1786-1793. http://dx.doi.org/10.1128/AEM.68.4.1786-1793.2002
- Smith, M.S., Yang, R.K., Knapp, C.W., Niu, Y.F., Peak, N., Hanfelt, M.M., Galland, J.C. and Graham, D.W. (2004) Quantification of Tetracycline Resistance Genes in Feedlot Lagoons by Real-Time PCR. Applied and Environmental Microbiology, 70, 7372-7377. http://dx.doi.org/10.1128/AEM.70.12.7372-7377.2004
- Aminov, R.I. and Mackie, R.I. (2007) Evolution and Ecology of Antibiotic Resistance Genes. FEMS Microbiology Letters, 271, 147-161. http://dx.doi.org/10.1111/j.1574-6968.2007.00757.x
- Peak, N., Knapp, C.W., Yang, R.K., Hanfelt, M.M., Smith, M.S., Aga, D.S. and Graham, D.W. (2007) Abundance of Six Tetracycline Resistance Genes in Wastewater Lagoons at Cattle Feedlots with Different Antibiotic Use Strategies. Environmental Microbiology, 9, 143-151. http://dx.doi.org/10.1111/j.1462-2920.2006.01123.x
- Knapp, C.W., Zhang, W., Sturm, B.S.M. and Graham, D.W. (2010) Differential Fate of Erythromycin and Beta-Lactam Resistance Genes from Swine Lagoon Waste under Different Aquatic Conditions. Environmental Pollution, 158, 1506-1512. http://dx.doi.org/10.1016/j.envpol.2009.12.020
- Chopra, I. and Roberts, M. (2001) Tetracycline Antibiotics: Mode of Action, Applications, Molecular Biology, and Epidemiology of Bacterial Resistance. Microbiology and Molecular Biology Reviews, 65, 232-260. http://dx.doi.org/10.1128/MMBR.65.2.232-260.2001
- Stanton, T.B., Humphrey, S.B., Scott, K.P. and Flint, H.J. (2005) Hybrid tet Genes and tet Gene Nomenculture: Request for Opinion. Antimicrobial Agents and Chemotherapy, 49, 1265-1266. http://dx.doi.org/10.1128/AAC.49.3.1265-1266.2005
- Levy, S.B., McMurry, L.M. and Roberts, M.C. (2005) Tet Protein Hybrids. Antimicrobial Agents and Chemotherapy, 49, 3099. http://dx.doi.org/10.1128/AAC.49.7.3099.2005
- Roberts, M.C. (2013) Tet Mechanisms of Resistance. http://faculty.washington.edu/marilynr/tetweb1.pdf
- Roberts, M.C., Sutcliffe, J., Courvalin, P., Jensen, L.B., Rood, J. and Seppala, H. (1999) Nomenclature for Macrolide and Macrolide-Lincosamide-Streptogramin B Resistance Determinants. Antimicrobial Agents and Chemotherapy, 43, 2823-2830.
- Chen, J., Yu, Z., Michel Jr., F.C., Wittum, T. and Morrison, M. (2007) Development and Application of Real-Time PCR Assays for Quantification of erm Genes Conferring Resistance to Macrolides-Lincosamides-Streptogramin B in Livestock Manure and Manure Management Systems. Applied and Environmental Microbiology, 73, 4407-4416. http://dx.doi.org/10.1128/AEM.02799-06
- Roberts, M.C. (2008) Update on Macrolide-Lincosamide-Streptogramin, Ketolide, and Oxazolidinone Resistance Genes. FEMS Microbiology Letters, 282, 147-159. http://dx.doi.org/10.1111/j.1574-6968.2008.01145.x
- Roberts, M.C. (2013) MLS Aquired Genes. http://faculty.washington.edu/marilynr/ermwebA.pdf
- Huys, G., D’Haene, K., Van Eldere, J., von Holy, A. and Swings, J. (2005) Molecular Diversity and Characterization of Tetracycline-Resistant Staphylococcus aureus Isolates from a Poultry Processing Plant. Applied and Environmental Microbiology, 71, 574-579. http://dx.doi.org/10.1128/AEM.71.1.574-579.2005
- Yu, Z., Michel Jr., F.C., Hansen, G., Wittum, T. and Morrison, M. (2005) Development and Application of Real-Time PCR Assays for Quantification of Genes Encoding Tetracycline Resistance. Applied and Environmental Microbiology, 71, 6926-6933. http://dx.doi.org/10.1128/AEM.71.11.6926-6933.2005
- Sharma, R., Larney, F.J., Chen, J., Yanke, L.J., Morrison, M., Topp, E., McAllister, T.A. and Yu, Z. (2009) Selected Antimicrobial Resistance during Composting of Manure from Cattle Administered Sub-Therapeutic Antimicrobials. Journal of Environmental Quality, 38, 567-575. http://dx.doi.org/10.2134/jeq2007.0638
- Melville, C.M., Scott, K.P., Mercer, D.K. and Flint, H.J. (2001) Novel Tetracycline Resistance Gene, tet(32), in the Clostridium-Related Human Colonic Anaerobe K10 and Its Transmission in Vitro to the Rumen Anaerobe Butyrivibrio fibrisolvens. Antimicrobial Agents and Chemotherapy, 45, 3246-3249. http://dx.doi.org/10.1128/AAC.45.11.3246-3249.2001
- Whittle, G., Whitehead, T.R., Hamburger, N., Shoemaker, N.B., Cotta, M.A. and Salyers, A.A. (2003) Identification of a New Ribosomal Protection Type of Tetracycline Resistance Gene, tet(36), from Swine Manure Pits. Applied and Environmental Microbiology, 69, 4151-4158. http://dx.doi.org/10.1128/AEM.69.7.4151-4158.2003
- Abril, C., Brodard, I. and Perreten, V. (2010) Two Novel Antibiotic Resistance Genes, tet(44) and ant(6)-Ib, Are Located within a Transferable Pathogenicity Island on Campylobacter fetus subsp. fetus. Antimicrobial Agents and Chemotherapy, 54, 3052-3055. http://dx.doi.org/10.1128/AAC.00304-10
- Wang, L., Oda, Y., Grewal, S., Morrison, M., Michel Jr., F.C. and Yu, Z. (2012) Persistence of Resistance to Erythromycin and Tetracycline in Swine Manure during Simulated Composting and Lagoon Treatments. Microbial Ecology, 63, 32-40. http://dx.doi.org/10.1007/s00248-011-9921-9
- Yu, Z. and Morrison, M. (2004) Improved Extraction of PCR-Quality Community DNA from Digesta and Fecal Samples. BioTechniques, 36, 808-812.
- Thompson, J.D., Gibson, T.J., Plewniak, F., Jeanmougin, F. and Higgins, D.G. (1997) The CLUSTAL_X Windows Interface: Flexible Strategies for Multiple Sequence Alignment Aided by Quality Analysis Tools. Nucleic Acids Research, 25, 4876-4882. http://dx.doi.org/10.1093/nar/25.24.4876
- Lozupone, C. and Knight, R. (2005) UniFrac: A New Phylogenetic Method for Comparing Microbial Communities. Applied and Environmental Microbiology, 71, 8228-8235. http://dx.doi.org/10.1128/AEM.71.12.8228-8235.2005
- Becker, S., Boger, P., Oehlmann, R. and Ernst, A. (2000) PCR Bias in Ecological Analysis: A Case Study for Quantitative Taq Nuclease Assays in Analyses of Microbial Communities. Applied and Environmental Microbiology, 66, 4945-4953. http://dx.doi.org/10.1128/AEM.66.11.4945-4953.2000

*Corresponding author.