Advances in Microbiology
Vol.4 No.9(2014), Article ID:48056,12 pages
DOI:10.4236/aim.2014.49059
Studies on Two Exoenzymes Which Lyse Wine-Spoiling Bacteria
Patrick Sebastian, Harald Claus*, Helmut König
Institute for Microbiology and Wine Research, Johannes Gutenberg University of Mainz, Mainz, Germany
Email: *hclaus@uni-mainz.de
Copyright © 2014 by authors and Scientific Research Publishing Inc.
This work is licensed under the Creative Commons Attribution International License (CC BY).
http://creativecommons.org/licenses/by/4.0/



Received 13 May 2014; revised 15 June 2014; accepted 15 July 2014
ABSTRACT
Microorganisms play an important role in the conversion of grape juice into wine. Different species of yeast are mainly responsible for the production of ethanol. Lactic acid bacteria also occur regularly in must or wine. They are mostly undesirable due to their capacity to produce winespoiling compounds. Especially strains of Lactobacillus brevis are able to produce biogenic amines as well as precursors of ethyl carbamate and different off-flavours (N-heterocycles, volatile phenols). By excessive formation of acetic acid some lactobacilli may even induce slow/stuck grape juice fermentations. In conventional winemaking, additions of sulphite or lysozyme are used to inhibit the growth of spoilage microorganisms. There is a strong interest in finding alternatives, because of the reduced activity of lysozyme in the wine milieu, a limited growth-inhibiting activity against lactic acid bacteria, and some health risks described regarding the application of sulphite. We found that a culture supernatant of Streptomyces albidoflavus B 578 lysed all bacteria previously isolated from must and wine samples (Acetobacter sp., Lactobacillus sp., Leuconostoc sp., Oenococcus oeni, Pediococcus sp., Staphylococcus sp.) including 35 strains of L. brevis. Two bacteriolytic exoenzymes were isolated and characterized from the streptomycete: a muramidase (24 kDa) and a protease (17 kDa). Both hydrolyzed cell wall components of L. brevis (peptidoglycan, S-layer proteins) and were active under wine-relevant conditions.
Keywords: Streptomyces albidoflavus, Lactobacillus brevis, Wine Spoilage, Stuck Fermentations, Cell Wall, Muramidase, Protease, S-Layer

1. Introduction
Yeast, lactic acid and acetic acid bacteria are the most abundant microbes occurring in must and wine [1] . In addition to positive metabolic activities, such as the alcoholic fermentation by yeasts or the malolactic fermentation by some lactic acid bacteria, these microbes are able to produce a wide range of metabolites which reduce the quality of wine [2] . In particular, strains of Lactobacillus brevis have been frequently isolated from must and wine [3] -[5] . They are spoiling microorganisms due to their ability to produce biogenic amines [5] -[7] , ethyl carbamate [6] , volatile phenols [8] and mousy off-flavors [9] .
The wine-maker conventionally uses sulphite to inhibit the growth of these spoilage organisms [10] . Unfortunately, the presence of sulphites in alcoholic beverages, particularly in wines, can cause pseudo-allergic reactions with symptoms ranging from gastrointestinal problems to anaphylactic shock [11] . The application of lysozyme, isolated from hen egg-white, has been allowed by the EU (EC amending regulation No 1622/2000) to control microbial growth during wine-making since 2001. Lysozyme is a cell-wall hydrolase which attacks the peptidoglycan layer of bacteria. The efficiency of the lytic activity depends on the peptidoglycan structure of the cell wall. Modifications, such as O-acetylation, N-deacetylation or the binding of teichonic acids to the peptidoglycan layer [12] , decrease the lytic activity of hen egg-white lysozyme. The occurrence of an outer membrane in Gram-negative bacteria or an S-layer in Gram-positive bacteria may further constrict the enzymatic accessibility of murein. The harsh physical conditions during wine-making (low pH values, high concentrations of ethanol and sulphite) also reduce the activity of this cell-wall hydrolase. Considering the fact that also hypersensitivity reactions to lysozyme have been reported [13] , it would be desirable to find alternative enzymes to inhibit wine-spoiling bacteria more efficiently.
Blättel et al. [14] reported on a lytic enzyme cocktail from Streptomyces sp. B 578 which is able to inhibit bacterial growth under wine-making conditions. In this study, the purification, identification and characterization of two enzymes from the bacteriolytic culture supernatant are described.
2. Materials and Methods
2.1. Bacterial Strains and Culture Conditions
Bacterial strains tested for sensitivity towards lytic enzymes have been isolated from must and wine samples and were identified by molecular methods [5] -[7] . Lactic acid bacteria were cultured in MRS medium [15] at 30˚C for two to three days. Acetic acid bacteria were grown in YPM broth (DSMZ medium no. 368; http://www.dsmz.de) on a shaker (100 rpm) at 30˚C for two days. LB medium (DSMZ medium no. 1) was used for the cultivation of the Gram-positive Micrococcus luteus B 577. Streptomyces albidoflavus B 578 was grown in GYM medium (DSMZ medium no. 65) under shaking (100 rpm) at 30˚C for two to three days. Species identification of strains B 577 and B 578 was performed by PCR amplification and sequencing of the 16S rRNA. The partial 1021 bp sequence of strain B 577 showed 100% identity with Micrococcus luteus strain SC1204 in the NCBI data base. Blast research with the complete 1534 bp sequence of strain B 578 (Ez-Taxon tool http://www.eztaxon.org) revealed 99.9% identity with the type strain Streptomyces albidoflavus DSM 40455. All microorganisms are deposited at the local culture collection of the Institute of Microbiology and Wine Research, Johannes Gutenberg-University Mainz.
2.2. Production of Bacteriolytic Exoenzymes
In order to produce lytic exoenzymes, S. albidoflavus B 578 was grown in 2 l flasks with 500 ml synthetic NMMP-medium [16] under moderate shaking (50 rpm) at 30˚C for six days. After cultivation, the supernatant was separated from the cells by centrifugation (8600 × g, 30 min). The culture supernatant was concentrated by ultrafiltration and dialyzed (cut-off: 3.5 kDa) against different buffers, depending on further experiments.
2.3. Determination of Bacteriolytic Activities
Bacteria to be tested for sensitivity against lytic enzymes were grown in the appropriate medium (MRS or YPM) to an approximate OD600 of 1.0 and washed twice with 10 mM Na+/K+ phosphate buffer (pH 5.9). The pellet was resuspended in 10 mM Na+/K+ phosphate buffer (pH 5.9) to obtain an OD600 of about 0.7. Quantification of the lytic activities was performed with a spectrophotometric test [17] . An amount of 50 µl of culture filtrate or pure enzyme solution were added to 1950 µl of the bacterial suspension and incubated at 30˚C for 4 h and/or 24 h. Afterwards, the suspensions were homogenized by shaking and the lytic activities determined as the percentage decrease of OD600 in comparison to a control assay without enzyme. All data presented are the mean values of triplicate determinations.
2.4. Protein Determination
Protein concentrations of the culture filtrates and pure enzyme solutions were determined with a BC (bicinchoninic acid) assay kit (Uptima, France).
2.5. Electrophoretic Methods
Protein compositions in the culture filtrates and fractions of the different purification steps were checked by SDS-PAGE [18] . Samples were separated in 10% (w/v) SDS gels (10 × 10 cm) with a 4% (w/v) stacking gel at room temperature for 1 h. A prestained protein ladder (PageRuler Plus, Fermentas, Germany) served as the molecular mass standard. Isoelectric focusing was carried out with ServalytPrecotes 3 - 10, following the recommendations of the manufacturer (Serva, Germany). Protein staining of SDS-PAGE and IEF gels was accomplished with Coomassie Blue R250, Serva Violet 17 (Serva, Germany) or by using a Silver-Staining Kit (Fermentas, Germany).
2.6. Purification of Lytic Enzymes
Culture filtrate (300 ml) of S. albidoflavus B 578 was concentrated by ultrafiltration via a 3.5 kDa cut-off membrane (Amicon, USA). This concentrate (10 ml) was dialyzed against a 20 mM citrate/phosphate buffer (pH 4.0) and applied to a cation exchange chromatography column (MonoS™ HR 5/5, Pharmacia Biotech, Sweden). Elution of the protein fractions collected (1.0 ml) occurred in a linear NaCl gradient from 0 to 1 M at a flow rate of 1.0 ml·min−1. The active protein fractions (see below) were further purified by gel permeation chromatography (Sephadex G-75, GE Healthcare, Germany) using a phosphate buffer, pH 7.2, for elution. Purity of the protein fractions were tested by SDS-PAGE and silver staining.
2.7. Determination of Molecular Masses and Isoelectric Points (IEP)
The apparent molecular masses of the purified enzymes were deduced from a calibration curve by blotting the electrophoretic migration distance of the standard proteins in SDS-PAGE versus the log of their molecular masses. The molecular masses of native proteins were determined by size exclusion chromatography (Sephadex G-75) using aprotonin (6.5 kDa), cytochrome C (12.4 kDa) and anhydrase from bovine erythrocytes (29 kDa) as references. The isoelectric point was estimated by comparison with the location of standard proteins in IEF gels.
2.8. Identification of Lytic Enzymes
The N-terminal amino acid sequences of proteins blotted from SDS polyacrylamide gels to polyvinylidene fluoride (PVDF) membranes were determined by Edman degradation with a protein sequencer (model 494A Procise, Applied Biosystems, USA). Furthermore, proteins were identified by mass spectroscopy [19] . Briefly, the samples were separated by SDS-PAGE and subjected to an in-gel tryptic digestion procedure. Tryptic peptides were transferred into an autosampler vial for analysis via LC-MS/MS. The tryptic peptides were separated by capillary liquid chromatography (LC) with a Waters NanoAcquity UPLC system (Waters Corporation, Milford, MA, USA) on a 100 µm × 100 mm CSH C18 column. The tryptic peptides were analysed with a Waters Xevo G2 QTOF mass spectrometry system using a positive mode electrospray ion source (ESI) using a NanoLockSpray source (Waters Corporation, Milford, MA, USA). The resulting tandem mass spectrometry data were processed and searched by using Proteinlynx Global Server, ver. 2.5.2. (Waters Corporation, Milford, MA, USA).
2.9. Preparation of Cell Walls
Cell walls of Lactobacillus brevis B 190 and Micrococcus luteus B 577 were prepared following the method of Schleifer and Kandler [20] . Cells harvested from a liquid culture were washed four times with deionized water. Glass beads (Ø 0.3 mm) were added and the mixture treated in a homogenizer at 10˚C for 20 min (Vibrogen-Zellmühle VI4, Bühler, Germany). The cell fragments were separated from the glass beads by rinsing through a Buchner funnel with deionized water. The crude cell walls were centrifuged (37,000 × g, 4˚C, 20 min) and washed twice in deionized water. The sediment was suspended in a small volume of phosphate buffer (0.05 M, pH 7.8) containing 0.5 mg·ml−1 trypsin. A few drops of toluene were added to the suspension and incubated at 37˚C overnight. The toluene was removed with a pipette followed by a gentle, brief boiling of the suspension. Then the suspension was centrifuged as above, washed in deionized water and freeze-dried.
2.10. Detection of Muramidase Activities
Muramidase activities were measured by the quantification of reducing sugars exposed after enzymatic cleavage of the peptidoglycan. The determination was performed according to Miller [21] with 3,5-dinitrosalicylic acid as the reagent. Isolated cell walls (2.5 mg·ml−1 in 50 mM sodium acetate buffer pH 6.0) were incubated with purified enzyme solution (10 µg·ml−1) at 30˚C for 2 h for the test. Reagent solution was added and the assay heated at 100˚C for 15 min. After cooling to room temperature, colour intensity was measured in a spectrophotometer at 575 nm. The concentration of reducing groups of N-acetyl-muraminic acid was estimated from a calibration curve with glucose.
2.11. Determination of the Muramidase Cleaving Site
Cell-wall preparations (1.0 mg·ml−1) of M. luteus B 577 were incubated with 24 µg·ml−1 purified enzyme solution from S. albidoflavus B 578, lysozyme (Sigma, Germany) or water at 37˚C for 24 h. A muramic acid solution (1 mg·ml−1) was treated equally to the cell-wall samples as reference. Afterwards, 1 mg NaBH4 was added to 100 µl sample and incubated on ice for 1 h. In order to separate the residual NaBH4, the samples were mixed with acetic acid and methanol and the borate methyl ester formed was evaporated in a vacuum concentrater (Eppendorf, Hamburg, Germany). Samples were diluted in 100 µl 6 N HCl and hydrolyzed at 100˚C for 2 h. Residual HCl was evaporated and the samples were dissolved in 100 µl ultrapure water for HPAEC-PAD (high performance anion exchange chromatography with pulsed amperometric detection) analysis. Samples were injected onto an anion exchanger chromatography column (CarboPac MA1, Dionex, Germany) and eluted in a gradient of water and 1 M NaOH at a flow rate of 0.4 ml·min−1. The chemically reduced muramic acid served as the standard of muramitol.
2.12. Isolation of S-Layer Proteins
S-layer proteins were isolated as described by Dohm et al. [22] . The bacterial biomass from 500 ml cultures of L. brevis B 190 grown at 30˚C for two days was harvested by centrifugation (9000 × g, 4˚C) and washed twice with sterile deionized water. One gram of wet cells was extracted with 5.0 ml of 5 M LiCl on a shaker (200 rpm) for 2 h and dialyzed against deionized water at 4˚C. The water-insoluble S-layer suspension was centrifuged at 16,000 × g for 15 min and stored at −20˚C.
2.13. Detection of Protease Activities
Protease activities were detected by using an assay by Gifford et al. [23] . A 0.5% azocasein solution (Serva, Germany) in 50 mM Tris-HCl (pH 7.2) was incubated with the purified enzyme at 37˚C for 24 h. After incubation, 20% (w/v) trichloroacetic acid was added and the sample was centrifuged at 13,000 × g for 30 min. The supernatant was mixed with 1 M NaOH and the extinction of the sample was determined spectrophotometrically at 440 nm.
2.14. Activity under Wine-Making Conditions
Enzyme activities were tested under six different temperatures (20˚C, 25˚C, 30˚C, 37˚C, 40˚C, and 50˚C), seven ethanol concentrations (0%, 2%, 5%, 7%, 10%, 12%, and 15% v/v) and nine pH values (3.0, 3.5, 4.0, 4.5, 5.0, 5.5, 6.0, 7.0, and 8.0). Cell lysis was measured with the methods described above.
3. Results
3.1. Bacteriolytic Activity of the Culture Supernatant of S. albidoflavus B 578
A total of 69 bacterial isolates from must and wine samples were tested for lysis by the exoenzymes of S. albidoflavus B 578 (Table 1). About half the number of test strains were lysed to more than 50% and only four strains were resistant. All 35 strains of Lactobacillus brevis were sensitive, whereas Oenococcus oeni especially displayed strain-dependent differences. Besides typical wine-related lactobacilli and the Gram-negative A. tropicales, both Staphylococcus species were lysed to a different extent.
3.2. Purification and Identification of Two Bacteriolytic Exoenzymes
The first purification step of the bacteriolytic enzymes from the culture supernatant of S. albidoflavus B 578 was carried out by fast performance liquid chromatography (FPLC) using a cation exchange column. After the separation into different fractions, each of them was tested for their bacteriolytic activity against L. brevis B 190. The lytic activities were concentrated in two main peaks in fractions 12 and 17, which were eluted at ca. 0.25 and 0.35 M NaCl, respectively, as shown in Figure 1. SDS-PAGE of fraction 12 delivered a main band at 17 kDa and two weaker bands at ca. 23 and 40 kDa. A single protein of 24 kDa could be detected in fraction 17 (not shown). Following size exclusion chromatography of fractions 12 and 17, two pure enzyme solutions with molecular masses around 17 kDa and 24 kDa, respectively, were obtained (Figure 2). In the case of the smaller enzyme, a total cell lysis of 52.4% was achieved at 30˚C within 24 h. The second enzyme was able to lyse 49.9% of the target cells under the same conditions (not shown). Hen egg-white lysozyme, which was used for comparison, showed no significant bacteriolytic activity against L. brevis B 190.

Table 1. Enzymatic lysis of bacteria isolated from wine samples by the culture supernatant of S. albidoflavus B 578.
*Lysis of cells given as percent decrease of OD600 after 24 h at 30˚C.
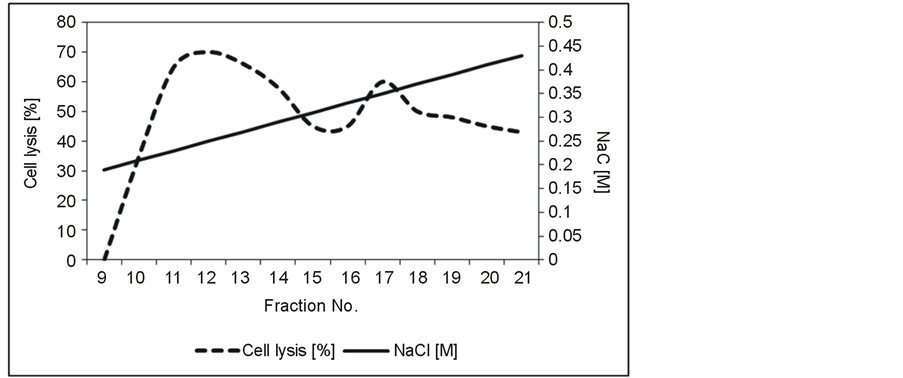
Figure 1. Elution profile of the culture supernatant from S. albidoflavus B 578 separated by cation exchange chromatography. Protein concentration applied: 0.29 mg·ml−1; elution buffer: 20 mM citrate buffer with 0.0 - 1.0 M NaCl; flow rate 1.0 ml·min−1. Cell lysis tests were performed with L. brevis strain B 190.
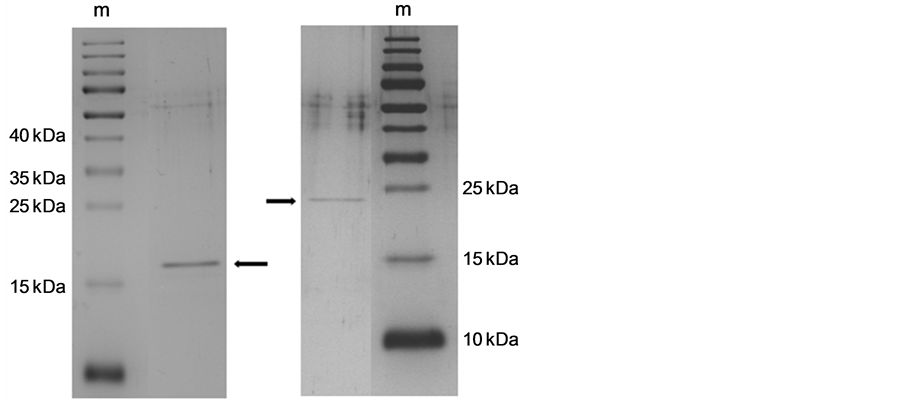
Figure 2. SDS-PAGE (15%) of the two purified bacteriolytic enzymes isolated from S. albidoflavus B 578. Left: muramidase (17 kDa); right: protease (24 kDa); m: marker proteins.
The purified enzymes were separated by SDS-PAGE, blotted onto PVDF membranes and sequenced by Edman degradation (Table 2). The N-terminus of the 17 kDa enzyme showed 100% similarity with trypsin-like serine endopeptidases of the streptogrisin B family firstly isolated from Streptomyces griseus. The sequence of the N-terminus for the 24 kDa enzyme was identical to different muramidases isolated from several species of Streptomyces. The results of Edman degradation were supported by mass spectroscopy of tryptic peptide fragments. By comparison with the reference spectra, the 17 kDa protein was identified as a streptogrisin B precursor, and the 24 kDa protein as a lysozyme M precursor, both found in the genome of Streptomyces albus J1074.
In accordance with the in silico data, the purified enzymes revealed proteolytic and glycanolytic activities, respectively (Figure 3). Their molecular masses and the isoelectric points were determined by SDS-PAGE and isoelectric focusing. The purified muramidase had a molecular mass of 23.4 kDa and an IEP of 8.8, whereas the protease had a molecular mass of 17.1 kDa and an IEP of 9.7. Nearly identical molecular masses (muramidase 24.9 kDa, protease 16.9 kDa) were deduced for the native enzymes from size exclusion chromatography (not shown), suggesting that the enzymes are active as monomers. The theoretical molecular masses of both enzymes from S. albus J 1074 calculated on the basis of the ORFs are considerably higher (Table 2). However, when calculated from the beginning of the N-termini, which are located within the sequences, there is good agreement with those of S. albidoflavus B 578. These results indicate that both lytic enzymes are activated by the enzymatic processing of precursor molecules.
3.3. Determination of the Substrate Cleavage Site of the Purified Cell-Wall Hydrolase
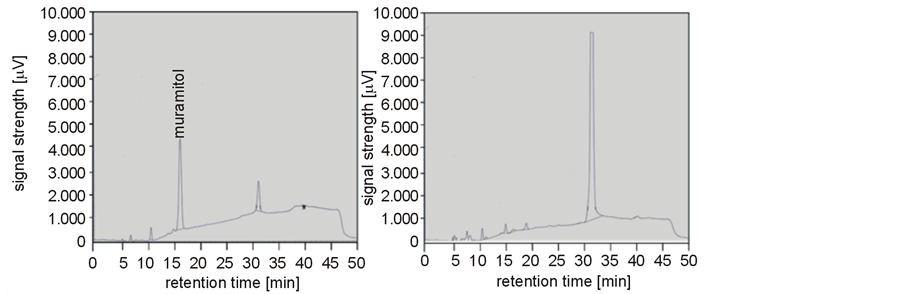
The enzymatic cleavage of cell-wall peptidoglycan by muramidase renders free carbonyl groups of the N-acetyl muramic acid. The sugar alcohol muramitol is produced by chemical reduction of these groups with NaBH4 and subsequent acid hydrolysis. This compound can be detected with anion exchange chromatography coupled with pulsed amperometric detection (HPAEC-PAD). Thus, a NaBH4-treated cell-wall preparation of M. luteus B577 was hydrolyzed and used as a negative control, and muramic acid treated equally as a positive control. In both cases, a signal with a retention time of 31 min could be detected (Figure 4(a), Figure 4(b)). This signal was significantly more pronounced in the sample with the cell wall, so it seems that this signal may present non-specific reaction products. Figure 4(a) shows a strong peak at about 16.5 min which belongs to muramitol. The cell-wall preparation was treated with lysozyme or the purified cell-wall hydrolase of S. albidoflavus B 578 for further analysis (Figure 4(c), Figure 4(d)). Both treatments resulted in the occurrence of a peak at 16.5 min. The release of muramitol in the sample after treatment with the enzyme confirms the muramidase activity, which cleaves the β-1,4-glycosidic bond between N-acetylmuramic acid and N-acetylglucosamine of the bacterial cell wall [24] .
Table 2. Results of the database research with the sequenced N-termini of the two purified enzymes from S. albidoflavus B 578*.
**Confirmed by mass spectrometrical identification of tryptic peptide fragments; aCalculated with the Protparam Tool (http://www.expasy.org); Properties of the putative mature enzymes beginning with the N-termini are given in brackets.
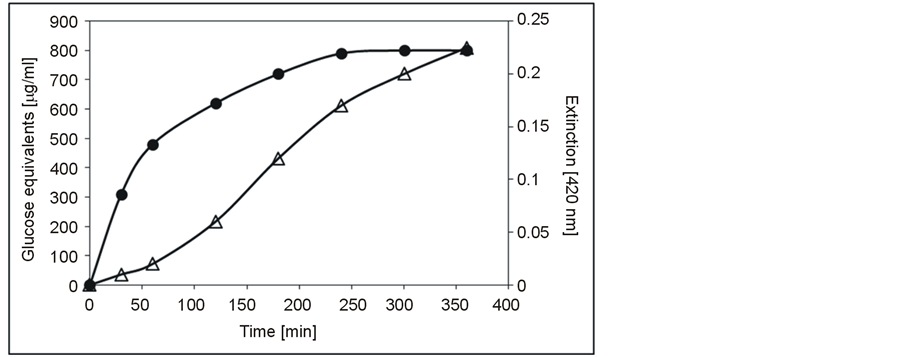
Figure 3. Detection of the proteolytic and cell-wall hydrolytic activities of the two purified exoenzymes of S. albidoflavus B 578 with the substrates azocasein (extinction Δ) and a cell-wall preparation of M. luteus B 577 (glucose equivalents ●).
3.4. Degradation of S-Layer Proteins by the Purified Protease
Isolated S-layer proteins were treated with the purified protease of S. albidoflavus B 578 and then separated by SDS-PAGE. Figure 5 shows the degradation kinetics of the prominent S-layer band at about 50 kDa and the appearance of non-specific lower molecular degradation products. Interestingly, the intensity of a new protein fragment at about 25 kDa (arrow) increased with the decrease of the S-layer protein. This molecular mass equals half of the purified S-layer. It could, therefore, be concluded that the S-layer protein consists of two equal-sized domains, and only one of those is sensitive against the protease of S. albidoflavus B 578. A similar result was found in our previous study for the S-layer protein from a Lactobacillus hilgardii strain [22] .
3.5. Bacteriolytic Activity under Wine-Relevant Conditions
It is important for the use of bacteriolytic enzymes in wine-making that they are active under the prevailing conditions in the wine milieu. Therefore, the activity of the purified enzymes was tested under different wine-relevant parameters. A bacteriolytic activity of both enzymes was given under all conditions tested, as shown in Table3 Regarding the pH value, the muramidase and the protease showed cell lysis of L. brevis B 190 at pH 3.0 and pH 3.5, respectively. These low pH values reflect the natural conditions of wine. No negative influence of
 (a)
(a)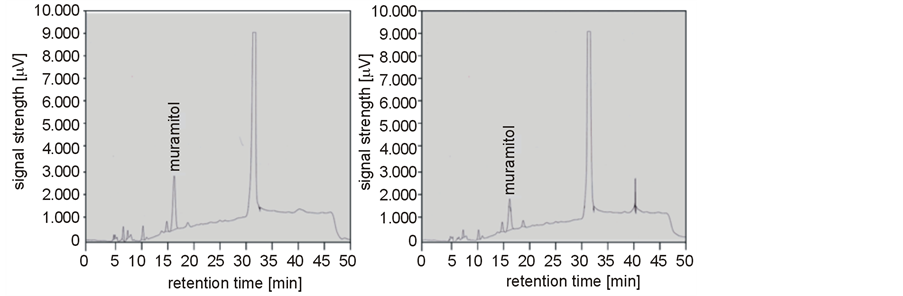 (b)
(b)
Figure 4. Detection of the muramolytic activity of the purified muramidase from S. albidoflavus B 578 by HPAEC-PAD. (a) Positive control (muramic acid, 1 mg·ml−1); (b) Negative control (cell wall of M. luteus B 577, 1 mg·ml−1); (c) Cell wall of M. luteus B 577 (1 mg·ml−1) treated with lysozyme (1 mg·ml-1); (d) Cell wall of M. luteus B 577 (1 mg·ml−1) treated with purified enzyme (24 µg·ml−1). Samples were obtained after chemical reduction and acid hydrolysis, as described in the material and methods section.
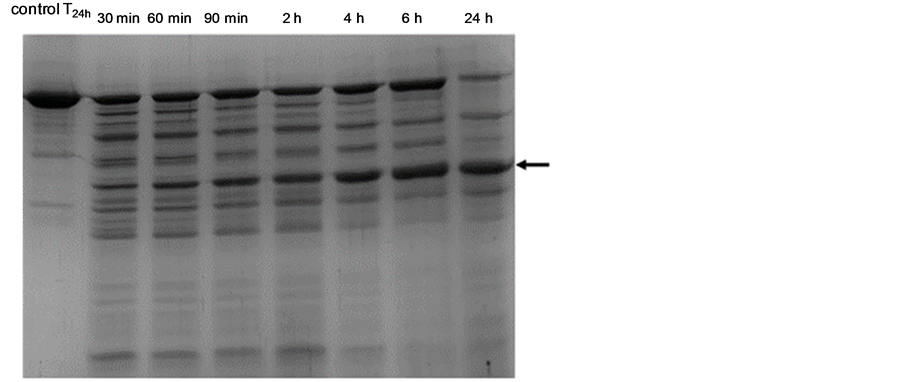
Figure 5. Degradation kinetics of the S-layer protein from L. brevis B190 by the purified protease from S. albidoflavus B 578 (77 µg·ml−1); the S-layer control contained no enzyme. Samples were taken at the given time intervals. The arrow indicates the major 25 kDa cleavage fragment.
Table 3. Bacteriolytic activity of the two purified enzymes from S. albidoflavus B 578 against L. brevis B 190 under different wine-related conditions.
Enzyme concentrations: protease 1.28 µg·ml−1, muramidase 0.72 µg·ml−1. Incubation time: 24 h at 30˚C*; 24 h at pH 7.0**; 24 h at 30˚C and pH 7.0***.
different ethanol concentrations on the activity was observed. The temperature of maximum enzymatic activity was measured at 30˚C. Some decrease of the activity occurred at higher temperatures. At lower temperatures, which are relevant for wine-making, the enzymes still showed high lysis rates against L. brevis B 190.
4. Discussion
We formerly demonstrated that Streptomyces sp. B 578, by now identified as a member of S. albidoflavus, produces a lytic enzyme cocktail useful for the control of wine-spoiling bacteria [14] . In the present study, a total of 66 out of 69 bacterial strains isolated from experimental wines were lysed by the culture supernatant of S. albidoflavus B 578. Strains of especially L. brevis, which have been identified as the most common producers of biogenic amines in wines [5] , could be lysed to a great extent. It was noticeable that O. oeni was lysed at lower percentages and in a strain-dependent manner. This is desirable due to the use of this bacterium as a starter culture for the implementation of the malolactic fermentation. By contrast, the widely used hen egg-white lysozyme has been reported to exert higher lytic activity against O. oeni strains than for the wine-spoiling Lactobacillus hilgardii [25] . Furthermore, lysis of the Gram-negative Acetobacter tropicalis was observed in our study. This is an advantage, because undesirable acetic acid bacteria have been found in young wine samples in increasing numbers in recent years [26] . In addition to wine-related microorganisms, bacteria of the genus Staphylococcus were lysed by the culture supernatant. As many staphylococci are known as human pathogens, the lytic enzymes of S. albidoflavus B 578 may also be of some medical interest [27] -[30] .
Cell-wall hydrolases are widely distributed among streptomycetes, e.g. in Streptomyces coelicolor [31] , Streptomyces globisporus [27] -[32] , Streptomyces griseus [33] , and Streptomyces rutgersensis [34] . They belong to the Chalaropsis type of lysozymes which display both N-acetylmuramidase and N,6-O-diacetylmuramidase activities. Cell-wall modifications in lactic acid bacteria, such as O-acetylations of N-acetylmuraminic acid, can restrict the activity of hen egg-white lysozyme [12] [35] [36] . The N,6-O-diacetymuramidase activity is probably responsible for the high bacteriolytic efficiency of S. albidoflavus B 578 against lactobacilli. Similar to our observation, the muramidase M1 from S. globisporus ATCC 21553 revealed a broad lysis spectrum against Gram-positive bacteria [37] . The muramidase of S. albidoflavus B 578 showed bacteriolytic activity at pH 3.0 to pH 8.0, ethanol concentrations of 0% to 15% (v/v) and within a temperature range of 12˚C to 50˚C. Thus, the activity of the enzyme could be proved in wine under general conditions.
The purified protease from S. albidoflavus B 578 showed a close phylogenetic relationship with the enzyme streptogrisin B found in several streptomycetes. This serine protease is a member of the chymotrypsin superfamily and is formed as part of the hydrolytic culture extract of Streptomyces griseus, which is referred to as pronase [38] . This protease is very similar to trypsin and splits particularly into phenylalanine-, tyrosine-, tryptophan-, and leucine-glycine bonds. Streptogrisin B was first described in S. griseus [39] and its 1.2 Å crystal structure presented by Lee and James [40] .
It was noticeable that the molecular masses calculated from the reference nucleotide sequences of the Streptomyces proteases published in the databases (Table 2) were far above the value of 17 kDa estimated from SDS-PAGE and gel filtration for the protease of our strain. However, when calculating the mass starting with the N-terminus obtained by Edman degradation, the experimental values could be confirmed. The active protease is most probably generated by cleavage of larger precursor molecules. The S. griseus protease C, for instance, has a pre-pro-mature organisation. The pre-region comprises the signal peptide for secretion (40 amino acids), followed by the propeptide of 162 amino acids, which is essential for the correct folding of the active protease (255 amino acids). The maturation of bacterial chymotrypsin-like proteases involves two proteolytic cleavages behind the preand the pro-region [38] .
In addition to the proteolytic activity against the common substrate azocasein, a bacteriolytic activity of the purified protease was demonstrated. Lysis and killing of bacteria by various proteases has been reported, however, the mode of action is not well understood. Some experimental data give hints that the proteases cleave peptide bonds in the peptidoglycan or the outer membrane of Gram-negative bacteria [41] . Another possible target are S-layer proteins, which comprise the outer cell border of many Archaea and Gram-positive bacteria, including lactic acid bacteria [22] [42] [43] . In this study, we found that the purified protease degrades S-layer proteins from L. brevis B 190, and previously, we demonstrated that the S-layer of Lactobacillus hilgardii B706 was degraded by the culture supernatant of S. albidoflavus B 578 [22] . L. hilgardii cells devoid of the S-layer are rendered more sensitive to bacteriolytic enzymes (e.g. achromopeptidase from Lysibacter) and wine-related stress conditions. From a practical point of view, the S-layer may be considered as a target for proteases to inhibit food-spoiling lactobacilli. As a prerequisite, activity of the protease from S. albidoflavus B 578 was confirmed under wine-making conditions. Above all, a pH optimum at pH 4.0 makes this enzyme interesting for wine-making. In addition, the wine industry is currently intensively searching for suitable proteases to prevent wine turbidity and to reduce possible allergenic proteins [44] .
5. Conclusion
In conclusion, our findings underline that bacteriolytic exoenzymes of S. albidoflavus B 578 could be useful in wine-making in order to inhibit lactobacilli responsible for wine spoilage [2] and stuck fermentations [45] . As both the protease and the muramidase displayed a broad lysis spectrum and high stability, their simultaneous use may support a synergistic action. Apart from the wine industry, general applications for microbial food control or medical purposes seem feasible.
Acknowledgements
The authors thank Bernhard Schlott (Leibniz-Institute for Age Research, Fritz Lipmann Institute, Jena, Germany) for identification of the N-termini by the Edman procedure, and Stefan Tenzer (Research Centre Immunology, FZI, of the Johannes Gutenberg-University, Mainz, Germany) for protein identification by mass spectroscopic methods. We appreciate the financial support by the Deutsche Forschungsgemeinschaft (DFG; Ko 785/17-1) and the FEI (Forschungskreis der Ernährungsindustrie E.V. Bonn) Projekt AIF-FV 15833N.
References
- Konig, H., Unden, G. and Frohlich, J. (2009) Biology of Microorganisms on Grapes, in Must and in Wine. Springer-Verlag, Heidelberg.
- Bartowsky, E.J. (2009) Bacterial Spoilage of Wine and Approaches to Minimize It. Letters in Applied Microbiology, 48, 149-156. http://dx.doi.org/10.1111/j.1472-765X.2008.02505.x
- Rodas, A.M., Ferrer, S. and Pardo, I. (2005) Polyphasic Study of Wine Lactobacillus Strains: Taxonomic Implications. International Journal of Systematic and Evolutionary Microbiology, 55, 197-207. http://dx.doi.org/10.1099/ijs.0.63249-0
- Dicks, L.M.T. and Endo, A. (2009) Taxonomic Status of Lactic Acid Bacteria in Wine and Key Characteristics to Differentiate Species. South African Journal of Enology and Viticulture, 30, 72-90.
- Sebastian, P., Herr, P., Fischer, U. and Konig, H. (2011) Molecular Identification of Lactic Bacteria Occurring in Must and Wine. South African Journal of Enology and Viticulture, 32, 300-309.
- Araque, I., Bordons, A. and Reguant, C. (2013) Effect of Ethanol and Low pH on Citrulline and Ornithine Excretion and arc Gene Expression by Strains of Lactobacillus brevis and Pediococcus pentosaceus. Food Microbiology, 33, 107-113. http://dx.doi.org/10.1016/j.fm.2012.09.005
- Petri, A., Pfannebecker, J.,
Frohlich, J. and Konig, H. (2013) Fast Identification of Wine Related Lactic Acid
Bacteria by Multiplex PCR. Food Microbiology, 33, 48-54.
http://dx.doi.org/10.1016/j.fm.2012.08.011 - Couto, J.A., Campos, F.M., Figueiredo, A.R. and Hogg, T.A. (2006) Ability of Lactic Acid Bacteria to Produce Volatile Phenols. American Journal of Enology and Viticulture, 57, 166-171.
- Costello, P.J., Lee, T.H. and Henschke, P.A. (2001) Ability of Lactic Acid Bacteria to Produce N-Heterocycles Causing Mausy Off-Flavour in Wine. Australian Journal of Grape and Wine Research, 7, 160-167. http://dx.doi.org/10.1111/j.1755-0238.2001.tb00205.x
- Ribéreau-Gayon, P., Glories, Y., Maujean, A. and Dubourdieu, D. (2006) Handbook of Enology Vol. 2—The Chemistry of Wine Stabilization and Treatments. 2nd Edition, John Wiley & Sons, Ltd., Hoboken. http://dx.doi.org/10.1002/0470010398
- Lin, S.C. and Georgiou, G. (2005) A Biocatalyst for the Removal of Sulfite from Alcoholic Beverages. Biotechnology and Bioengineering, 89, 123-127. http://dx.doi.org/10.1002/bit.20307
- Logardt, I.M. and Neujahr, H.Y. (1975) Lysis of Modified Walls of Lactobacillus fermentum. Journal of Bacteriology, 124, 73-77.
- Pérez-Calderón, R., Gonzalo-Garijo, M.A., Lamilla-Yerga, A., Mangas-Santos, R. and Morena-Gastón, I. (2007) Recurrent Angiodema Due to Lysozyme Allergy. Journal of Investigational Allergology and Clinical Immunology, 17, 264-266.
- Blattel, V., Wirth, K., Claus, H., Schlott, B., Pfeiffer, P. and Konig, H. (2009) A Lytic Cocktail from Streptomyces sp. B578 for the Control of Lactic and Acetic Acid Bacterial in Wine. Applied Microbiology and Biotechnology, 83, 839-848. http://dx.doi.org/10.1007/s00253-009-1926-7
- De Man, J.C., Rogosa, M. and Shape, M.R. (1960) A Medium for the Cultivation of Lactobacilli. Journal of Applied Bacteriology, 23, 130-135. http://dx.doi.org/10.1111/j.1365-2672.1960.tb00188.x
- Hopwood, D.A., Bibb, M.J., Chater, K.F., Kieder, T., Bruton, C.J., Kieser, H.M., Lydiate, D.J., Smith, C.P., Ward, J.M. and Schrempf, H. (1985) Genetic Manipulation of Streptomycetes—A Laboratory Manual. The John Innes Foundation, Norwich.
- Ohbuchi, K., Hasegawa, K., Hamachi, M., Ozeki, K. and Kumagai, C. (2001) Isolation of a New Lytic Enzyme for Hiochi Bacteria and Other Lactic Acid Bacteria. Journal of Bioscience and Bioengineering, 91, 487-492.
- Laemmli, U.K. (1970) Cleavage of Structural Proteins during the Assembly of the Head of Bacteriophage T4. Nature, 227, 680-685. http://dx.doi.org/10.1038/227680a0
- Claus, H., Tenzer, S., Sobe, M., Schlander, M., Konig, H. and Frohlich, J. (2014) Effect of Carboxymethyl Cellulose on Tartrate Salt, Protein and Colour Stability of Red Wine. Australian Journal of Grape and Wine Research, 20, 186-193. http://dx.doi.org/10.1111/ajgw.12070
- Schleifer, K.H. and Kandler, O. (1972) Peptidoglycan Types of Bacterial Cell Walls and Their Taxonomic Implications. Bacteriological Reviews, 36, 407-477.
- Miller, G.L. (1959) Use of Dinitrosalicylic Acid Reagent for Determination of Reducing Sugar. Analytical Chemistry, 31, 426-428. http://dx.doi.org/10.1021/ac60147a030
- Dohm, N., Petri, A., Schlander, M., Schlott, B., Konig, H. and Claus, H. (2011) Molecular and Biochemical Properties of the S-Layer Protein from the Wine Bacterium Lactobacillus hilgardii B706. Archives of Microbiology, 193, 251-261. http://dx.doi.org/10.1007/s00203-010-0670-9
- Gifford, A., Klippenstein, J.
and Moore, M. (2002) Serum Stimulates Growth of and Proteinase Secretion by Aspergillus
fuminatus. Infection and Immunity, 70, 19-26.
http://dx.doi.org/10.1128/IAI.70.1.19-26.2002 - Vollmer, W., Joris, B., Charlier, P. and Foster, S. (2008) Bacterial Peptidoglycan (Murein) Hydrolases. FEMS Micro-biological Reviews, 32, 259-286. http://dx.doi.org/10.1111/j.1574-6976.2007.00099.x
- Delfini, C., Cersosimo,
M., Del Prete, V., Strano, M., Gaetano, G., Pagliara, A. and Ambró, S. (2004) Resistance
Screening Assay of Wine Lactic Acid Bacteria on Lysozyme: Efficacy of Lysozyme in
Unclarified Musts. Journal of Agricultural and Food Chemistry, 52, 1861-1866.
http://dx.doi.org/10.1021/jf034824m - Wirth, K., Sebastian, P., Schneider, I. and Konig, H. (2011) Vorkommen von Essigsaurebakterien in Jungweinen: Isolierung neuer Arten. Der Deutsche Weinbau, 6, 36-37.
- Yokogawa, K., Kawata, S. and Yoshimura, Y. (1973) Studies on Lytic Enzyme against Cariogenic Streptococci. II. Lytic Enzyme from Streptomyces globisporus 1829 Strain. Agricultural and Biological Chemistry, 37, 799-808. http://dx.doi.org/10.1271/bbb1961.37.799
- Salazar, O. and Asenjo, J.A. (2007) Enzymatic Lysis of Microbial Cells. Biotechnological Letters, 29, 984-994. http://dx.doi.org/10.1007/s10529-007-9345-2
- Callewaert, L., Walmagh, M.,
Michiels, C.W. and Livigne, R. (2011) Food Applications of Bacterial Cell-Wall Hydrolases.
Current Opinions in Biotechnology, 22, 164-171.
http://dx.doi.org/10.1016/j.copbio.2010.10.012 - Szweda, P., Schielmann, M., Kotlowski, R., Gorczyca, G., Zalewska, M. and Milewski, S. (2012) Peptidoglycan Hydrolases—Potential Weapons against Staphylococcus aureus. Applied Microbiology and Biotechnology, 96, 1157-1174. http://dx.doi.org/10.1007/s00253-012-4484-3
- Rau, A., Hogg, T., Marquardt, R. and Hilgenfeld, R. (2001) A New Lysozyme Fold. Crystal Structure of the Muramidase from Streptomyces coelicolor at 1.65 A. Journal of Biological Chemistry, 276, 31994-31999. http://dx.doi.org/10.1074/jbc.M102591200
- Seo, H.J., Shimonishi, T., Ohmiya, K. and Hayashi, K. (2001) Characterization of N-acetylmuramidase M-1 of Streptomyces globisporus Produced by Escherichia coli BL21 (DE3) pLysS. Journal of Bioscience and Bioengineering, 92, 472-474.
- Yokogawa,
K., Kawata, S. and Yoshimura, Y. (1976) Purification and Properties of a Lytic Enzyme
from Streptomyces griseus H-402. Agricultural and Biological Chemistry, 40, 661-667.
http://dx.doi.org/10.1271/bbb1961.40.661 - Hayashi, K., Kasumi, T., Kubo, N. and Tsumura, N. (1981) Purification and Characterization of the Lytic Enzyme Produced by Streptomyces rutgersensis H-46. Agricultural and Biological Chemistry, 45, 2289-2300. http://dx.doi.org/10.1271/bbb1961.45.2289
- Clarke, A. and Dupont, C. (1992)
O-Acetylated Peptidoglycan: Its Occurrence, Pathobiological Significance and Biosynthesis.
Canadian Journal of Microbiology, 38, 85-91.
http://dx.doi.org/10.1139/m92-014 -
Delcour, J., Ferain, T., Deghorain, M., Palumbo, E. and Hols, P. (1999) The Biosynthesis
and Functionality of the Cell-Wall of Lactic Bacteria. Antonie van Leeuwenhoek,
76, 159-184.
http://dx.doi.org/10.1023/A:1002089722581 - Seo, H.J., Kitaoka, M., Ohmiya, K. and Hayashi, K. (2003) Substrate Specificity of the N,6-O-Diacetylmuramidase from Streptomyces globisporus. Journal of Bioscience and Bioengineering, 95, 313-316. http://dx.doi.org/10.1016/S1389-1723(03)80037-3
- Sidhu, S.S., Kalmar, G.B., Willis, L.G. and Borgford, T.J. (1994) Streptomyces griseus Protease C. Journal of Biological Chemistry, 269, 20167-20171.
- Wahlby, S. and Engstrom, L. (1968) Studies on Streptomyces griseus Protease: II. The Amino Acid Sequence around the Reactive Serine Residue of DFP-Sensitive Components with Esterase Activity. Biochimica et Biophysica Acta, 151, 402-408. http://dx.doi.org/10.1016/0005-2744(68)90107-1
- Lee, T.W. and James, M.N.G. (2008) 1.2Angstrom-Resolution Crystal Structures Reveal the Second Tetrahedral Intermediates of Streptogrisin B (SGPB). Biochimica et Biophysica Acta (BBA)-Proteins and Proteomics, 1784, 319-334. http://dx.doi.org/10.1016/j.bbapap.2007.11.012
- Thorne, K.J., Oliver, R.C. and Barrett, A.J. (1976) Lysis and Killing of Bacteria by Lysosomal Proteinases. Infection and Immunity, 14, 555-563.
- Claus, H., Akca, E., Debaerdemaeker, T., Evrard, C., Declecq, J.P., Harris, J.R., Schlott, B. and Konig, H. (2005) Molecular Organization of Selected Prokaryotic S-Layer Proteins. Canadian Journal of Microbiology, 51, 731-743. http://dx.doi.org/10.1139/w05-093
- Hynonen, U. and Palva, A. (2013) Lactobacillus
Surface Layer Proteins: Structure, Function and Applications. Applied Microbiology
and Biotechnology, 97, 5225-5243.
http://dx.doi.org/10.1007/s00253-013-4962-2 -
Waters, E.J. and Colby, C.B. (2009) Proteins. In: Morena-Arribas, M.V. and Polo,
M.C., Eds., Wine Chemistry and Biochemistry, Springer Science & Business Media,
New York, 213-230.
http://dx.doi.org/10.1007/978-0-387-74118-5_11 - Edwards, C.G., Reynolds, A.G., Rodriguez, A.V., Semon, M.J. and Mills, J.M. (1999) Implication of Acetic Acid in the Induction of Slow/Stuck Grape Juice Fermentations and Inhibition of Yeast by Lactobacillus sp. American Journal of Enology and Viticulture, 50, 204-210.
NOTES

*Corresponding author.