Advances in Microbiology
Vol.3 No.3(2013), Article ID:33780,7 pages DOI:10.4236/aim.2013.33039
Development of a Microplate Lectin-Capture RT-PCR (MLC-RT-PCR) for the Detection of Avian Infectious Bronchitis Virus
1Laboratory of Virology and Immunology—IMUNOVIR—Department of Veterinary Pathology of University of Sao Paulo State (FCAV-UNESP), Jaboticabal, Brazil
2Brazilian National Research Center on Swine and Poultry (CNPSA-EMBRAPA), Concordia, Brazil
3Department of Preventive Veterinary Medicine and Animal Health, Veterinary Medicine School of Sao Paulo University (FMVZ-USP), São Paulo, Brazil
Email: *mariafsm@fcav.unesp.br
Copyright © 2013 Maria de Fatima S. Montassier et al. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Received December 2, 2012; revised January 17, 2013; accepted February 17, 2013
Keywords: Molecular Diagnosis; Infectious Bronchitis Virus; RNA Separation; Lectin-Capture
ABSTRACT
Rapid, sensitive and specific methods are necessary to confirm the diagnosis of outbreaks of avian infectious bronchitis virus (IBV) infection. The amplification of IBV genome by reverse transcription followed by polymerase chain reaction (RT-PCR) has been one of the most used methods for the detection of this virus in clinical samples. To reduce the time and the number of steps in the molecular diagnosis of IBV, we developed a sensitive and rapid detection method based on viral capture by a lectin (Concanavalin A—Con A) in the microplate wells, followed by RT-PCR to amplify the S1 gene. The detection limit of IBV was 103 EID50/ml for the amplification of 5’part of the S1 gene, and 104 EID50/ml for the amplification of full S1 gene. This technique was specific for IBV detection, and no amplified products were detected for other avian viral pathogens (bursal infectious disease virus, avian metapneumovirus and Newcastle disease virus). The MLC-RT-PCR was as sensitive as conventional RT-PCR, and virus isolation method for the detection of IBV in tissue samples collected from experimentally infected birds. The MLC-RT-PCR technique demonstrated a great potential for the rapid and specific diagnosis of IBV.
1. Introduction
Infectious bronchitis virus (IBV) is one of the major causes of economic losses in the poultry industry and can be result in respiratory disease, nephritis, and poor egg production and egg quality [1,2]. However, these pathological signs of the disease are not specific to IBV [1,2]. IBV is globally distributed and extremely difficult to control [1-4]. Therefore, fast, sensitive and specific virus detection techniques are of great interest to the poultry health program in order to identify IBV infections in the field. Identification may also include typing of the isolate involved in order to define the most effective vaccination program affording sufficient protection against IBV infection [3,4].
IBV is an important avian pathogen characterized by a worldwide distribution and many different variants of this virus are appearing continuously, despite the use of vaccines [4]. This virus is classified in the Gammacoronavirus Genus from the Coronaviridae Family [5]. The IBV contains a genome constituted by a single stranded RNA of positive polarity that consists of approximately 27 kb and codes for four main structural proteins: the spike glycoprotein (S), the small envelope (E) protein, the membrane glycoprotein (M) and the nucleocapside protein (N) [6].
Although virus isolation in embryonated chicken eggs remaining the gold standard for IBV direct diagnosis [7], molecular diagnosis based on different RT-PCR techniques has been increasingly used for the detection of parts of IBV genome from clinical samples. This is due to relevant advantages over virus isolation, especially because the molecular diagnostic techniques can be combined with RFLP and nucleic acid sequencing to genotype the IBV isolates [3].
The quality of the extracted RNA is critical for the success of RT-PCR, especially the removal of RT-PCR inhibitors from the clinical samples. Viral RNA has usually been extracted from these samples by various commercial kits, which are based on the columns containing resins, or alternatively on the use of magnetic beads coated with silica or other substances, for adsorption and elution of nucleic acid [8,9]. Conventional and real-time RT-PCR methods reported for the detection of the IBV genome have been done with viral RNA extracted from allantoic fluid, swab and tissue samples by different methods, but the most used methods were TRIzol/modification of the single-step acid guanidium thiocyanatephenol-chloroform extraction technique, column kits and magnetic bead kits [10-16]. However, these methods have some drawbacks for RNA separation, including the need for special equipments, the handling of hazardous chemicals, and high costs.
Immunocapture procedures via polyclonal or monoclonal anti-viral antibodies have been extensively used for the attachment of virus particles in a solid phase (plastic microplates, tubes, or magnetic beads), followed by viral RNA elution for using in RT-PCR [9, 17-20]. However, to date there have been no reports on using this or other types of capture methods for the immobilization of IBV followed by the extraction of viral RNA.
A number of lectins isolated from plants and invertebrates have been evaluated as capturing reagents when adsorbed to solid phase supports, such as magnetic beads, for the attachment of gram-positive and gram-negative bacteria from food samples. This procedure resulted in the concentration of the target bacteria and reduced the presence of contaminants, contributing to the isolation of these bacteria [21].
Based on the binding capacity of Con A with mannose moieties of viral glycoproteins, this lectin has been used for the separation of RNA viruses, as togaviruses, influenzavirus, parainfluenzavirus, and rhabdoviruses [22]. Interactions between mannose-binding lectins, other than Con A, and coronaviruses (SARS-coronavirus and feline infectious peritonitis virus) were detected and resulted in antivirus activity, due to interference at virus entry and virus release [23]. Additionally, Con A captured efficiently IBV in the ELISA microplates [24], but there are no reports describing the use of lectins to immobilize and separate Coronaviruses from crude viral suspensions for use in molecular diagnostic techniques. Thus, as the lectin Con A binds to terminal mannose groups of oligo-saccharides presented by S and M glycoproteins of IBV [25], a MLC-RT-PCR was developed based on the PCR amplification of cDNA synthesized from the viral RNA of lectin-captured viruses.
In this study, a RT-PCR coupled with a lectin-capture method are described, using microplate wells as solidsupport for the detection of IBV in allantoic fluid and tissue samples.
2. Materials and Methods
2.1. Viruses
One reference strain (M41) and 5 Brazilian field isolates of IBV were tested. These isolates were previously sequenced and S1-genotyped [26,27]. The GenBank accession numbers for S1 nucleotide sequences were GQ169239 (IBVPR01), GQ169246 (IBVSC01), GQ169247 (IBVSC02), GQ169243 (IBVPR06), GQ169250 (IBVSP02). These viruses were propagated in 9 to 10-day-old specific pathogen free (SPF) embryonated chicken eggs [28]. The eggs were inoculated by the allantoic sac route, and the IBV infected allantoic fluid suspensions were collected after 36 hs. The embryo 50% infectious doses (EID50) were determined [29] for M41 strain and IBVSC01 isolate. The IB viruses were kindly provided by Empresa Brasileira de Pesquisa Agropecuária-EMBRAPA (Concordia-SC, Brazil).
2.2. MLC-RT-PCR
Polystyrene microtitre plates (96-well plates) were coated with 200 ml of 0.5 mg/ml of Concanavalin A (Sigma Chemical Co., St. Louis, MO), diluted in PBS pH 7.4 and incubated overnight at 4˚C. One well out of four was used, in order to avoid cross-contamination. The wells were washed four times with PBS. After washing, 200 ml of virus samples were added and the reaction was incubated for 1 h at 37˚C. The wells were washed six times with PBS, taking care to avoid any cross-contamination. The reverse transcription (RT) was carried out in the same plate, by adding to each well 20 ml of the RT mixture containing 200 U of SuperScript II reverse transcriptase (Invitrogen, Carlsbad, CA) and 25 pmol of random hexamers as primer (Invitrogen, Carlsbad, CA). The plate was incubated for 1 h at 42˚C. The whole volume of each well containing cDNA solution, was transferred to 0.2 ml polypropylene tubes and stored at −20˚C. Two set of primers were used for PCR; one (primer set 1) is specific for the amplification of the 5’part of S1 gene [30], and the other was specific for the amplification of the entire S1 gene [31]. The reaction was performed in volumes of 50 ml, containing 5 ml of the cDNA, 20 pmol of each specific primer and 2.0 U Taq DNA polymerase (Invitrogen, Carlsbad, CA). The PCR techniques were conducted as described [30,31] for 5’part or the entire S1 gene, respectively. PCR products were analyzed on a 1% agarose gel containing (0.5 mg/ mL) of ethidium bromide.
2.3. Conventional RT-PCR for IBV Detection in Tissue Samples
The same procedure described above for LC-RT-PCR was adopted, except that the RNA was extracted using TRIzol LS reagent (Invitrogen, Carlsbad, CA), according to the manufacturer’s instructions.
2.4. Analytical Specificity of MLC-RT-PCR
The analytical specificity of the LC-RT-PCR was assessed using different antigen suspensions of the M41 strain of IBV and IBV Brazilian field isolates (IBVPR01, IBVPR03, IBVPR06, IBVSC02, IBVSP02), as well as non-related viruses such as Newcastle Disease Virus (NDV/La Sota vaccine strain), avian pneumovirus (AVP PL-21 vaccine strain) and infectious bursal disease virus (IBDV/Lukert vaccine strain).
2.5. Analytical Sensitivity of MLC-RT-PCR
A series of ten-fold dilutions (100 to 10−7) of a stock IBV strain (M41), where the virus infectivity determined in embryonated chicken eggs corresponded to 10−7/ml embryo 50% embryo infectious doses (EID50/ml), were tested on the LC-RT-PCR, as described above. The analytical sensitivity corresponded to the highest dilution that gave a specific amplified product.
2.6. Tissue Samples for IBV Detection
Three groups of 15 specific pathogen free (SPF) White Leghorn chickens were maintained in positive pressure isolators units. At four weeks of age, the birds of the first group were inoculated with 106 EID50 of M41 strain of IBV, and the birds of the second group were inoculated with 106 EID50 of a Brazilian field isolate (IBVSC01). A third group remained non-infected in another isolator unit and served as the negative control. Tracheal samples were collected from each group 5 days post-infection. Tissue samples were tested for virus isolation (Viral Isol.) in embryonated chicken eggs, [32]. The tissue samples were considered negative or positive on virus isolation based on the absence or the presence of typical embryonic lesions induced by IBV, such as stunting, curling or embryo death [32]. These tissue specimens were also submitted to MLC-RT-PCR, conventional RT-PCR using total RNA extracted by TRIZOL, and to Con A-SELISA.
2.7. Con A-S-ELISA
The Con A-Sandwich ELISA was carried out according to [24] for the detection of IBV antigen in allantoic fluid or in tissue samples.
3. Results
The IBV was effectively captured by the lectin Con A attached to the microplate wells, and after the addition of the buffer and RT reagents generated, after appropriate incubation, a cDNA which was amplified in the PCR, by primer sets 1 and 2 for S1 gene (Figure 1).
The specificity of the MLC-RT-PCR was demonstrated for either the primer set-1 or the primer set-2. Amplified products with 452 bp and 1.7 kb were only detected in allantoic fluid samples infected with one reference strain (M41), or one of the five field isolates of IBV (Figure 1). None of the other viruses non-related to IBV (Newcastle disease virus, avian metapneumovirus, infectious bursal disease virus) tested here showed any one of these amplified products (Figure 1).
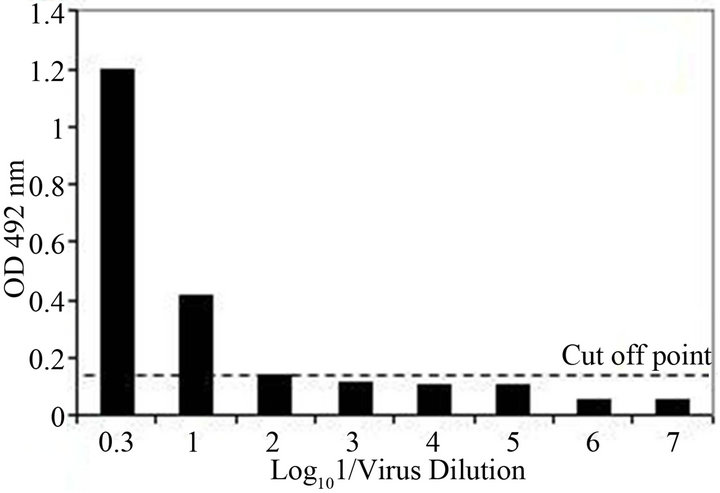
The analytical sensitivity of the MLC-RT-PCR was assessed for ten-fold dilutions of AF infected with M41 strain of IBV, using the primer sets 1 or 2 and the results are shown in Figure 2. After gel electrophoresis a 452 bp band, for primer set-1, was detected down to the dilution of 10−4 (103 DIE50), and a 1.7 kb band, for primer set-2, was visible at the dilution of 10−3 (104 DIE50). The Con A–S–ELISA exhibited a lower analytical sensitivity and IBV was detected down to 10−2 dilution (Figure 2). The same sensitivity was obtained by conventional RT-PCR, using primer sets 1 or 2 (Figure 3), and assaying the viral RNA extracted by TRIzol LS reagent (Invitrogen, Carlsbad, CA).
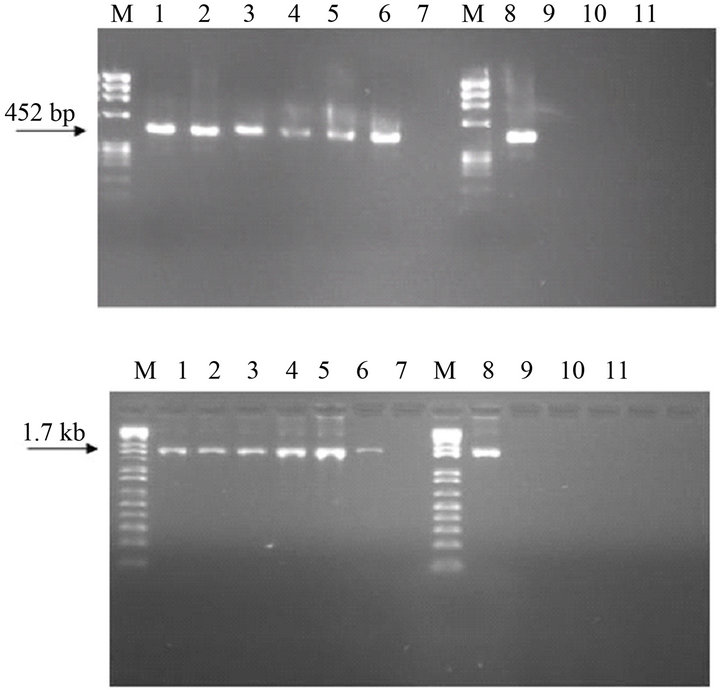
Figure 1. Specificity analysis of LC-RT-PCR, using SYUprimers (amplicon of 452 bp) or the Oligo-S1-primers (amplicon of 1.7 kb). M-DNA marker 1 kb plus, Lane 1—IBVM41, Lane 2—IBVPR01, Lane 3—IBVSC01, lane 4— IBVSC02, Lane 5—IBVPR06, Lane 6—IBVSP02, Lane 7— negative control, Lane 8—IBVM41, Lane 9—avian metapneumovirus, Lane 10—Newcastle disease virus, Lane 11— infectious bursal disease virus, Lane 12—negative control.
 (a)
(a)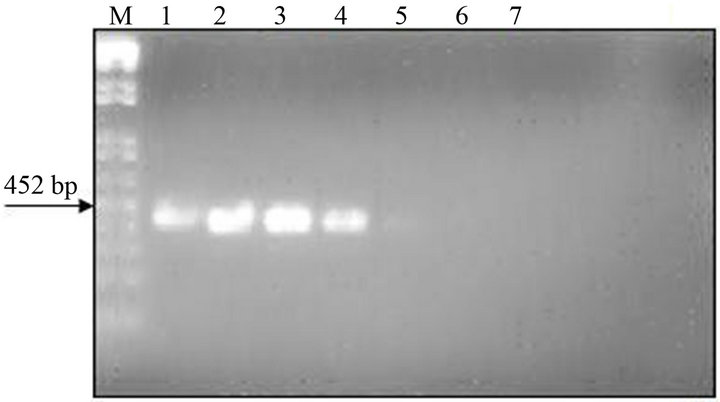 (b)
(b) (c)
(c)
Figure 2. Sensitivity of Con A-S-ELISA and LC-RT-PCR for the detection of IBV (M41 strain) present in AF suspendsion. Panel (a): Con A-S-ELISA optical density values. Panel (b): Agarose gel electrophoresis analysis of RT-PCR (SYU-primers) products of 5’part of S1 gene of IBV (M41 strain). Panel (c): Agarose gel electrophoresis analysis of RT-PCR (S-Oligo-1-primers) products of entire S1 gene of IBV (M41 strain). M-DNA marker 1 kb (Invitrogen). Lane 1—undiluted IBV suspension, Lane 2 to 6—ten-fold dilutions of IBV suspension, starting 10−1 through 10−6 and Lane 7—negative virus suspension.
The MLC-RT-PCR, using primer set-1, detected 80 to 100% of the tracheal samples, collected in acute phase from chickens experimentally infected with M41 strain, or with IBVSC01 isolate, respectively (Table 1). There was a complete agreement between the results of MLCRT-PCR and the conventional RT-PCR for the detection of the IBV genome in samples from both experimentally infected groups. The virus isolation in embryo-
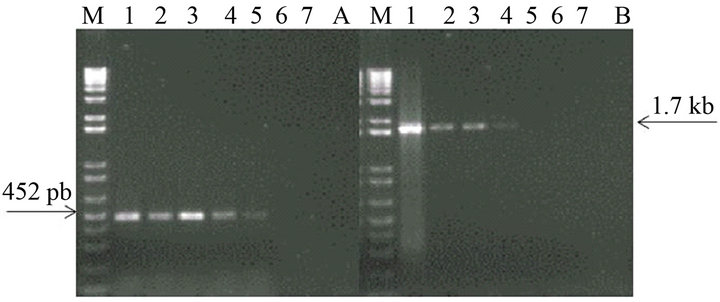
Figure 3. Sensitivity of conventional RT-PCR (RNA extraction by TRIzol LS reagent) for the detection of IBV (M41 strain) present in AF suspension. Panel A: Agarose gel electrophoresis analysis of RT-PCR (SYU–primers) products of 5’part of S1 gene of IBV (M41 strain). Panel B: Agarose gel electrophoresis analysis of RT-PCR (S-Oligo-1- primers) products of entire S1 gene of IBV (M41 strain). M–DNA marker 1 kb (Invitrogen). Lane 1—undiluted IBV suspension, Lane 2 to 6—ten-fold dilutions of IBV suspension, starting 10−1 through 10−6 and Lane 7—negative virus suspension.
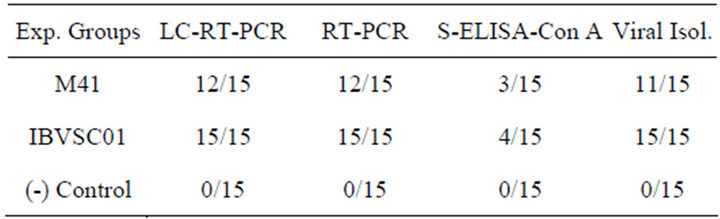
Table 1. Comparison of IBV detection in tissue samples collected from experimentally infected and non-infected birds by different diagnostic methods.
nated chicken eggs detected 73.33% to 93.33% of tracheal samples, collected from the groups of experimentally infected chickens, while 20 to 26.66% of these samples were detected by Con A-S-ELISA. No positivity was detected in the tissue samples from the negative control group.
4. Discussion
Conventional IBV diagnosis is quite complex and labor-intensive, because several passages in embryonated SPF chicken eggs are usually required before field isolates can produce typical lesions in the embryos [1-4]. The molecular diagnostic methods, though they have demonstrated efficiency, face some limitations, particularly with regard to the extraction of viral RNA.
To overcome these difficulties, an alternative molecular biology method that combines a virus capture to a solid phase mediated by the lectin Con A and RT-PCR technique was developed for the detection of IBV in allantoic fluid, or in tissue samples collected from experimentally infected eggs or chickens, respectively. This method is designed so that the entire procedure, includeing the RT step, can be carried out for each sample in the same microplate well. A similar procedure has been applied to detect rabbit haemorrhagic disease virus and European brown hare syndrome virus [18]. MLC-RTPCR does not require any previous handling of samples for extraction of viral RNA, or even a thermal disruption of trapped virions to release the genomic RNA.
The MLC-RT-PCR using primer set-1, and based on the infectivity titer (EID50/ml) of the viral suspension tested, was as sensitive for the detection of ten-fold dilution series of IBV-infected AF samples as the conventional RT-PCR technique previously developed with the N-gene primer set [33], and it was more sensitive than a conventional RT-PCR with S1 primers [33]. Additionally, the sensitivity of MLC-RT-PCR with primer set-1 was in the range of those reported for RT-PCR involving RNA extraction by conventional methods and the detection of other RNA viruses, as well as IBV [18,33-35]. The MLC-RT-PCR using primer set-1 was more sensitive than the reaction with primer set-2, probably because the PCR product amplified by primer set-1 is smaller (452 bp) than that amplified by primer set-2 (1.7 kb). In comparison with ELISA, the MLC-RT-PCR was 100 to 1000 times more sensitive than the Con A-S-ELISA. Similar sensitivity was found for the detection of plant viruses [36,37], or animal viruses, such as foot-and-mouth disease virus and European brown hare syndrome virus, when compared to ELISA [18,34].
Six IB viruses, including the M41 reference strain, and five Brazilian field isolates, classified, or in the Massachusetts genotype (one isolate), or in variant genotypes (four isolates) [26,27], were effectively detected by the MLC-RT-PCR. This indicates that the mannoside oligosaccharides present in S and M glycoproteins of Massachusetts strain are well conserved among different IBV isolates, since these carbohydrate motifs are critical for the binding of these viruses to Con A [24,25]. In addition, although mannose could be present in the glycoproteins of other avian viruses, including Paramyxoviruses [22], which might lead to the capture of viruses non-related to IBV, the specificity of this method remained high, because it depends directly on the pair of primers used in the PCR. Thus, heterologous viruses, such as avian metapneumovirus, Newcastle disease virus and infectious bursal disease virus were not detected by the MLCRT-PCR, although the interactions of Con A with molecular components of these viruses can not be ruled out.
A high sensitivity was observed for the detection of IBV in tissue samples from experimentally infected birds by comparing MLC-RT-PCR with conventional RT-PCR, or with the virus isolation method in embryonated SPF chicken eggs. The detection range of IBV in tissue samples by MLC-RT-PCR is comparable to those obtained by the conventional and real time RT-PCR techniques previously applied [11,33].
From this study, it can be assumed that MLC-RT-PCR can be advantageously applied for IBV detection in clinical samples, because it is simpler, appropriate for processing a large number of samples, and takes less time than virus isolation, which requires a minimum of three egg passages to confirm the IBV diagnosis. Furthermore, MLC-RT-PCR has some advantages when compared to conventional RT-PCR performed with total RNA extracted by organic solvents or column kits, because it does not need special devices or equipment and it is less expensive. Additionally, microplates, or other solid phase supports (microtubes, magnetic beads) adsorbed with Con A could be prepared and stored, giving the lectin-capture of IBV for viral RNA separation a similar length of time to complete.
In conclusion, the Con A efficiently captured the IBV particles and permitted the elution of viral RNA to be amplified by RT-PCR. In addition, MLC-RT-PCR was as effective as conventional RT-PCR performed with RNA extracted by TRIzol for the amplification of S1 gene in clinical samples, and can be applied to the molecular diagnosis of IBV infection.
5. Acknowledgements
The authors are grateful to Fundação de Amparo à Pesquisa no Estado de São Paulo (FAPESP), Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq for financial support) and MERIAL for providing SPF embryonated chicken eggs. M.F.S. Montassier was granted a PHD scholarship from FAPESP.
REFERENCES
- D. Cavanagh, “Coronavirus Avian Infectious Bronchitis Virus,” Veterinary Research, Vol. 38, No. 2, 2007, pp. 281-297. doi:10.1051/vetres:2006055
- D. Cavanagh and J. F. Gelb Jr., Infectious Bronchitis, In: Y. M. Saif, Ed., Diseases of Poultry, 12th Edition, WileyBlackwell, Hoboken, 2008, pp. 117-137.
- J. J. De Wit, “Detection of Infectious Bronchitis Virus,” Avian Pathology, Vol. 29, No. 2, 2000, pp. 71-93. doi:10.1080/03079450094108
- J. Ignjatović and S. Sapats, “Avian Infectious Bronchitis Virus,” Revue Scientifique et Technique, Vol. 19, No. 2, 2000, pp. 493-508.
- International Committee on Taxonomy of Viruses. http://ictvonline.org/virusTaxonomy.asp?version=2011
- M. M. C. Lai and D. Cavanagh, “The Molecular Biology of Coronaviruses,” Advances in Virus Research, Vol. 48, 1997, pp. 1-100. doi:10.1016/S0065-3527(08)60286-9
- World Organisation for Animal Health (OIE), Avian Infectious Bronchitis. Chapter 2.3.2. Manual of Diagnostic Tests and Vaccines for Terrestrial Animals, 6th Edition, OIE, Paris, 2008.
- M. Y. Deng, H. Wang, G. B. Ward, T. R. Beckham and T. S. McKenna, “Comparison of Six RNA Extraction Methods for the Detection of Classical Swine Fever Virus by Real-Time and Conventional Reverse Transcription-PCR” Journal of Veterinary Diagnostic Investigation, Vol. 17, No. 6, 2005, pp. 574-578. doi:10.1177/104063870501700609
- R. Dhumpa, K. J. Handberg, P. H. Jørgensen, S. Yi, A. Wolff and D. D. Bang, “Rapid Detection of Avian Influenza Virus in Chicken Fecal Samples by Immunomagnetic Capture Reverse Transcriptase-Polymerase Chain Reaction Assay,” Diagnostic, Microbiology and Infectious Disease, Vol. 69, No. 3, 2011, pp. 258-265. doi:10.1016/j.diagmicrobio.2010.09.022
- D. Cavanagh, K. Mawditt, P. Britton and C. J. Naylor, “Longitudinal Field Studies of Infectious Bronchitis Virus and Avian Pneumovirus in Broilers Using Type-Specific Polymerase Chain Reactions,” Avian Pathology, Vol. 28, No. 6, 1999, pp. 593-605. doi:10.1080/03079459994399
- K. J. Handberg, O. L. Nielsen, M. W. Pedersen and P. H. Jorgensen, “Detection and Strain Differentiation of Infectious Bronchitis Virus in Tracheal Tissue from Experimentally Infected Chickens by Reverse TranscriptionPolymerase Chain Reaction. Comparison with an Immunohistochemical Technique,” Avian Pathology, Vol. 28, No. 4, 1999, pp. 327-335. doi:10.1080/03079459994579
- M. W. Jackwood, D. A. Hilt and S. A. Callison, “Detection of Infectious Bronchitis Virus by Real-Time Reverse Transcriptase-Polymerase Chain Reaction and Identification of a Quasispecies in the Beaudette Strain,” Avian Diseases, Vol. 47, No. 3, 2003, pp. 718-724. doi:10.1637/6075
- C. H. Okino, M. F. S. Montassier, P. E .N. Givisiez, C. R. A. G. Furuyama, L. Brentano and H. J. Montassier, “Infectious Bronchitis Virus: Detection and Vaccine Strain Differentiation by Semi-Nested RT-PCR,” Brazilian Journal of Poultry Science, Vol.7, No. 1, 2005, pp. 59-66.
- S. A. Callison, D. A. Hilt, T. O. Boynton, B. F. Sample, R. Robison, D. E. Swayne and M. W. Jackwood, “Development and Evaluation of a Real-Time Taqman RT-PCR Assay for the Detection of Infectious Bronchitis Virus from Infected Chickens,” Journal of Virological Methods, Vol. 138, No. 1-2, 2006, pp. 60-65. doi:10.1016/j.jviromet.2006.07.018
- K. Hewson, A. H. Noormohammadi, J. M. Devlin, K. Mardani and J. Ignjatovic, “Rapid Detection and NonSubjective Characterisation of Infectious Bronchitis Virus Isolates Using High-Resolution Melt Curve Analysis and a Mathematical Model,” Archives of Virology, Vol. 154, No. 4, 2009, pp. 649-660. doi:10.1007/s00705-009-0357-1
- R. M. Jones, R. J. Ellis, W. J. Cox, J. Errington, C. Fuller, R. M. Irvine and P. R. Wakeley, “Development and Validation of RT-PCR Tests for the Detection and S1 Genotyping of Infectious Bronchitis Virus and Other Closely Related Gammacoronaviruses within Clinical Samples,” Transboundary and Emerging Diseases, Vol. 58, No. 5, 2011, pp. 411-420. doi:10.1111/j.1865-1682.2011.01222.x
- R. W. Jansen, G. Siegl and S. M. Lemon, “Molecular Epidemiology of Human Hepatitis A Virus Defined by an Antigen-Capture Polymerase Chain Reaction Method,” Proceedings of National Academy of Science, Vol. 87, No. 8, 1990, pp. 2867-2871. doi:10.1073/pnas.87.8.2867
- G. Le Gall-Reculé, F. Zwingestein, Y. Portejoie and G. Le Gall, “Immunocapture-RT-PCR Assay for Detection and Molecular Epidemiology Studies of Rabbit Haemorrhagic Disease and European Brown Hare Síndrome viruses,” Journal of Virological Methods, Vol. 97, No. 1-2, 2001, pp. 49-57. doi:10.1016/S0166-0934(01)00336-6
- Y. Park, Y. H. Cho, Y. Jee and G. Ko, “Immunomagnetic Separation Combined with Real-Time Reverse Transcriptase PCR Assays for Detection of Norovirus in Contaminated Food,” Applied and Environmental Microbiology, Vol. 74, No. 13, 2008, pp. 4226-4230. doi:10.1128/AEM.00013-08
- L. Yao, Q. Wu, D. Wang, X. Kou and J. Zhang, “Development of Monoclonal Antibody-Coated Immunomagnetic Beads for Separation and Detection of Norovirus (Genogroup II) in Faecal Extract Samples,” Letters in Applied Microbiology, Vol. 49, No. 2, 2009, pp. 173-178. doi:10.1111/j.1472-765X.2009.02638.x
- M. J. Payne. S. Campbel and R. G. Kroll, “Lectin-Magnetic Separation Can Enhance Methods for the Detection of Staphylococcus aureus, Salmonella enteritidis and Listeria monocytogenes,” Food Microbiology, Vol. 10, No. 1, 1993, pp. 75-83. doi:10.1006/fmic.1993.1008
- H. Becht, R. Rott and H. D. Klenk, “Effect of Concanavalin A on Cells Infected with Enveloped RNA Viruses,” Journal of General Virology, Vol. 14, No. 1, 1972, pp. 1- 8. doi:10.1099/0022-1317-14-1-1
- E. Keyaerts, L. Vijgen, C. Pannecouque, E. Van Damme, W. Peumans, H. Egberink, J. Balzarini and M. Van Ranst, “Plant Lectins Are Potent Inhibitors of Coronaviruses by Interfering with Two Targets in the Viral Replication Cycle,” Antiviral Research, Vol. 75, No. 3, 2007, pp. 179- 187. doi:10.1016/j.antiviral.2007.03.003
- R. R. V. M. Bronzoni, M. F. S. Montassier, G. T. Pereira, N. M. Q. Gama, V. Sakai and H. J. Montassier, “Detection of Infectious Bronchitis Virus and Specific Anti-Viral Antibodies Using a Concanavalin A-Sandwich-ELISA,” Viral Immunology, Vol. 18, No. 3, 2005, pp. 269-578. doi:10.1089/vim.2005.18.569
- J. A. Lancer and C. R. Howard, “The Polypeptides of Infectious Bronchitis Virus (IBV-41 strain),” Journal of General Virology, Vol. 46, No. 2, 1980, pp. 349-361. doi:10.1099/0022-1317-46-2-349
- M. F. S. Montassier , L. Brentano, L. J. Richtzenhain and H. J. Montassier, “Genetic Diversity on S1 Glycoprotein of Avian Infectious Bronchitis Virus Strains Isolated in Brazil between 1988-2000,” Vol. 1, Proceedings of the 5th International Symposium on Avian Coronavirus, Rauischholzhausen, 14-16 May 2006, pp. 119-131.
- M. F. S. Montassier , L. Brentano, H. J. Montassier and L. J. Richtzenhain, “Genetic Grouping of Avian Infectious Bronchitis Virus Isolated in Brazil Based on RT-PCR/ RFLP Analysis of the S1 Gene,” Pesquisa Veterinária Brasileira, Vol. 28, No. 3, 2008, pp. 190-194. doi:10.1590/S0100-736X2008000300011
- R. L. Owen, “Detection of Viral Antigen Following Exposure of One-Day-Old Chicken to the Holland-52-Strain of IBV,” Avian Pathology, Vol. 20, No. 4, 1991, pp. 663- 673. doi:10.1080/03079459108418805
- P. Villegas, “Titration of Biological Suspensions,” In: D. E. Swayne, J. R. Glisson, M. W. Jackwood, J. E. Pearson and W. M. Reed, Eds., A Laboratory Manual for the Isolation and Identification of Avian Pathogens, The American Association of Avian Pathologists, Pennsylvania, 1998, pp. 248-254.
- L. Yu, Z. Wang, Y. Jiang, S. Low and J. Kwang, “Molecular Epidemiology of Infectious Bronchitis Virus Isolates from China and Southeast Asia,” Avian Diseases, Vol. 45, No. 1, 2001, pp. 201-209. doi:10.2307/1593029
- H. M. Kwon, M. W. Jackwood and J. F. Gelb Jr., “Differentiation of Infectious Bronchitis Virus Serotypes Using Polymerase Chain Reaction and Restriction Fragment Length Polymorphism Analysis,” Avian Diseases, Vol. 37, No. 1, pp. 194-202. doi:10.2307/1591474
- R. E. Gough, D. J. Alexander, A. S. Collins and W. J. Cox, “Routine Virus Isolation or Detection in the Diagnosis of Diseases in Birds,” Avian Pathology, Vol. 17, No. 4, 1988, pp. 893-906. doi:10.1080/03079458808436511
- R. Meir, O. Maharat, Y. Farnushi and L. Simanov, “Development of a Real-Time TaqMan RT-PCR Assay for the Detection of Infectious Bronchitis Virus in Chickens, and Comparison of RT-PCR and Virus Isolation,” Journal of Virological Methods, Vol. 163, No. 2, 2010, pp. 190-194. doi:10.1016/j.jviromet.2009.09.014
- A. Rodriguez, J. I. Nuñez, G. Nolasco, F. A. Ponz, F. Sobrino and C. de Blas, “Direct PCR Detection of FootAnd-Mouth Disease Virus,” Journal of Virological Methods, Vol. 47, No. 3, 1994, pp. 345-349. doi:10.1016/0166-0934(94)90030-2
- C. L. Kho, M. L. Mohd-Azmi, S. S. Arshad and K. Yusoff, “Performance of an RT-Nested PCR ELISA for the Detection of Newcastle Disease Virus,” Journal of Virological Methods, Vol. 86, No. 1, 2001, pp. 71-83. doi:10.1016/S0166-0934(99)00185-8
- G. Nolasco, C. de Blas, V. Torres and F. A. Ponz, “A Method Combining Immunocapture and PCR Amplification in a Microtiter Plate for the Detection of Plant Viruses and Subviral Pathogens,” Journal of Virological Methods, Vol. 45, No. 2, 1993, pp. 201-218. doi:10.1016/0166-0934(93)90104-Y
- T. Wetzel, T. Candresse, G. Macquaire, M. Ravelonandro and J. Dunez, “A Highly Sensitive Immunocapture Polymerase Chain Reaction Method for Plum Pox Potyvirus Detection,” Journal of Virological Methods, Vol. 39, No. 1-2, 1992, pp. 27-37. doi:10.1016/0166-0934(92)90122-T
NOTES
*Corresponding author.