International Journal of Clinical Medicine
Vol.3 No.3(2012), Article ID:19386,4 pages DOI:10.4236/ijcm.2012.33038
A Patient with Intravenous Immunoglobulin-Responsive Lower Motor Neuron Syndrome
![]()
1Department of Internal Medicine, Hiratsuka City Hospital, Hiratsuka, Japan; 2Department of Neurology, Hiratsuka City Hospital, Hiratsuka, Japan; 3Department of Neurology, Kitasato University School of Medicine, Sagamihara, Japan; 4Department of Neurology, National Hakone Hospital, Odawara, Japan; 5Department of Neurology, Tokai University School of Medicine, Isehara, Japan.
Email: *yoshii@is.icc.u-tokai.ac.jp
Received January 9th, 2012; revised February 28th, 2012; accepted March 19th, 2012
Keywords: lower motor neuron syndrome (LMNS); multifocal motor neuropathy (MMN); localized proximal muscle weakness; intravenous immunoglobulin (IVIg); anti-GM1 ganglioside antibody
ABSTRACT
We report a 50-year-old woman who developed localized proximal muscle weakness, in addition to transient elevation of antibodies to GM1 ganglioside, without multifocal conduction block. She was treated with intravenous immunoglobulin (IVIg) and steroid pulse therapy, which were effective for over 10 years. Her clinical course and laboratory tests were consistent with lower motor neuron syndrome (LMNS) with localized proximal muscle weakness. We suggest that some patients diagnosed as LMNS may remain responsive to IVIg or steroid pulse therapy for a long time.
1. Introduction
Lower motor neuron syndrome (LMNS) should be distinguished from multifocal motor neuropathy (MMN), but the differential diagnosis is sometimes difficult. MMN is presumed to have an immune-mediated pathogenesis and is treated with steroid or intravenous immunoglobulin (IVIg). Asymmetric muscle weakness of the distal upper limbs with multifocal motor conduction block and a positive response to treatment with IVIg are considered the most relevant criteria. However, LMNS includes a heterogeneous group of diseases, and the distributions of muscle weakness may be predominantly asymmetrical or symmetrical, distal or proximal, and situated in arms or legs [1,2]. Progression may be rapid or only rapid initially followed by spontaneous arrest, or slow throughout the course. Patients are usually unresponsive to immunotherapy. Therefore, careful electrophysiological evaluation to search for conduction blocks is required. We describe an adult-onset LMNS patient who showed localized proximal muscle weakness, but had a positive response to treatment with IVIg or steroid pulse therapy for over 10 years.
2. Case Report
A 50-year-old woman had gait disturbance and weakness of the upper limbs, which appeared in 1998. She visited our hospital in August, 1999 for progressive muscle weakness. She had no relevant medical history except for sudden deafness, which had already improved. On examination, she showed limitation of shoulder muscle elevation and positive Gower’s sign. Muscle strength by mMRC (modified Medical Research Council) criteria was 2 in bilateral deltoid muscles, 4 in bilateral biceps brachii and triceps brachii muscles and 4 in bilateral quadriceps femoris and paravertebral muscles. However, the muscle strength of wrist and finger and tibialis anterior muscles was normal. Her sensation and tendon reflex were normal, and autonomic function tests were within normal ranges. Blood tests were normal except for mild elevation of creatinine kinase (216 IU/l). M-protein, antinuclear antibody, antineutrophil antibody, anti-SS-A and anti-SS-B antibodies were all negative. IgM anti-GM1 ganglioside antibody titer was mildly elevated (×400). Cerebrospinal fluid examination showed normal cell counts with total protein 49 mg/dl. Magnetic resonance imaging (MRI) presented marked atrophy and fatty degeneration of deltoid, quadriceps femoris and paravertebral muscles (Figure 1). A needle electromyogram showed neuropathic changes in quadriceps femoris, biceps femoris, and gastrocnemius muscles, but normal biceps brachii, interossei dorsalis, semimembranosus, and tibialis anterior muscles. Motor conduction velocities, distal latencies, and amplitudes of compound muscle action potential
(CMAP) were close to normal. A conduction block was observed on the segment of the right median nerve between axilla and elbow at the onset, but the finding was not confirmed subsequently. Muscle biopsy of biceps brachii muscle (Figure 2) did not show characteristic findings of myositis, because alkaline phosphatase (ALP) staining was negative both in muscle cells and connective tissues. A few small angulated fibers were stained with neuron specific enolase (NSE). Type1 fibers were dominant, and Type 2C fibers were intermediately scattered without fiber type grouping. These findings were consistent with mild neuropathic changes. The patient was treated with IVIg 0.4 g/kg for 5 days (November, 1999). Gower’s sign became negative, and the other muscle symptoms were somewhat improved. Two months later, she was treated with plasma exchange, which did not produce a clear improvement of muscle weakness. Then, steroid pulse therapy was performed with 1 g intravenous methylprednisolone for 3 days (July, 2000), followed by oral prednisolone (1 mg/kg, daily). After the treatment, her weakness in the upper limbs was improved, and the titer of anti-GM1 ganglioside antibody decreased and has remained negative since 2000. However, her symptoms, such as Gower’s sign or limitation of shoulder elevation reappeared, and steroid pulse therapy has been repeated periodically for 10 years. The intervals between steroid pulse therapy became gradually shorter, and the effects became less marked. On November 2009, because of osteoporosis as a side effect of steroid, oral prednisolone was discontinued, and she was again treated with IVIg therapy, which was effective. During 13-years’ follow-up, muscle weakness and atrophy has remained localized symmetrically at proximal muscles.
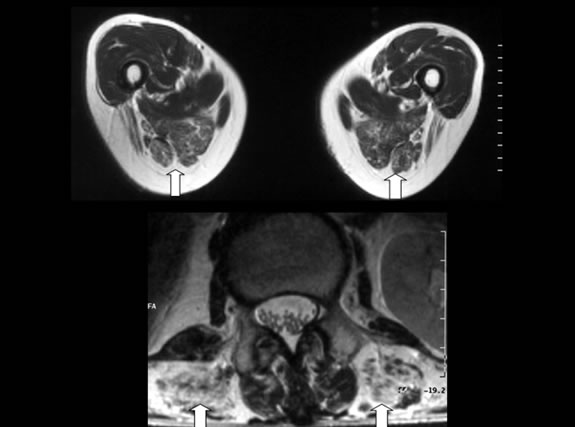
Figure 1. T1-weighted MRI axial images of lower limbs (upper) and paravertebral muscle (lower). Arrows: atrophic muscles.
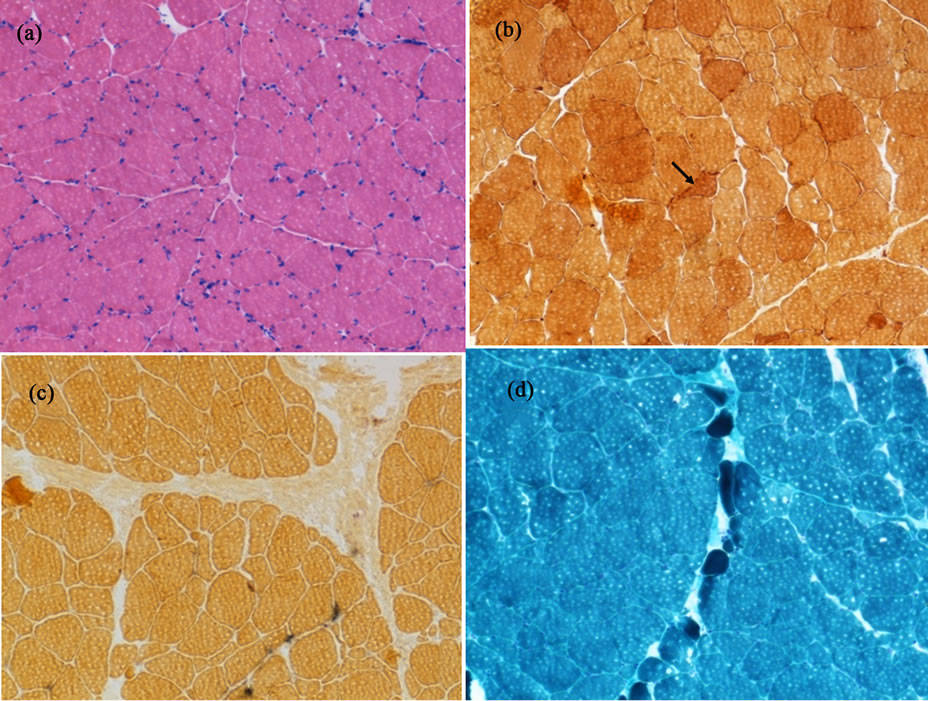
Figure 2. Muscle biopsy of biceps brachii muscle. (a) Hematoxylin and eosin staining; (b) Immunostaining using neuron specific enolase (arrow: small angulated fiber); (c) Immunostaining using alkaline phosphatase; (d) Gomori trichrome staining.
3. Discussion
The clinical features of LMNS, which closely resemble the progressive spinal muscular atrophy of adult motor neuron disease [3,4], can be subdivided into two or more forms based on clinical, physiological, and immunological criteria [5]. The most distinct form is seen in patients with MMN [6], in which nerve conduction block in at least one motor nerve is considered an essential finding [7]. However, since Pestronk described MMN without conduction block, other similar cases have been reported under names such as LMNS [8,9], MMN without overt conduction block [10], chronic motor axonal neuropathy (CMAN) [11,12], or axonal multifocal motor neuropathy without conduction block [13]. Electrophysiological studies cannot distinguish them from ALS. These cases often showed pure lower motor neuron involvement, in addition to the existence of anti-ganglioside antibodies (GM1 [8], GalNAc-GD1a [12]) and trivial evidence for demyelination in conduction studies [10]. Favorable response to treatment such as IVIg [9] was sometimes observed.
Our patient showed localized weakness in proximal muscles, such as deltoid, quadriceps femoris and paravertebral muscles, without sensory disturbance. A motor nerve conduction block was noted only once at one segment in the early stage. There was no remarkable delay of conduction velocity or temporal dispersion of CMAP, which does not satisfy the criteria for MMN. However, immune therapies such as IVIg or steroid were effective. Pestronk et al. described a similar patient with proximal LMNS [5], who showed proximal dominant weakness, without little elevation of antibodies to GM1. Proximal dominant LMNS patients in this series differed from patients with MMN, based on clinical, nerve conduction and immunological criteria. Clinically, these diseases are especially common in men and onset is often relatively late. The weakness was predominantly in proximal muscles and often remained within one or two extremities for at least 3 to 5 years. Conduction block in the proximal portion of the fibers was unusual. Serum tests often showed high titers of antibodies to asialo-GM1 and Galβ1-3GalNAc, but these were negative in our patient.
Recently Burrel et al. [14] reported that more than 70% of patients with pure LMNS responded to IVIg therapy, despite a low prevalence of detectable GM1 antibodies and conduction block. They noted that patients with predominantly upper limb, asymmetrical, and distal weakness were more likely to respond to IVIg therapy. However, our patient had symmetrical and proximal weakness. Thus, clinical symptoms might not be a good predictor of treatment response [15].
Our patient presented muscle atrophy and weakness in localized proximal parts without multifocal motor conduction block. Steroid pulse and IVIg treatments remained effective for over 10 years. She does not satisfy the criteria of MMN, and belongs to the category of LMNS. However, she showed transient elevation of IgM anti-GM1 gangliside antibody and conduction block on one segment of the right median nerve at the onset. These findings may easily be overlooked in the course of LMNS. So we suggest that it is necessary to review the effect of immune therapy, even in patients atypical for MMN.
4. Conclusion
Patients with LMNS may respond to IVIg or steroid pulse therapy, irrespective of the presence or absence of antiGM1 ganglioside antibodies or conduction block, and should be given an empirical trial of IVIg or steroid to determine treatment responsiveness. Our patient showed a good response without progressive symptoms for over 10 years.
REFERENCES
- R. M. Van Den Berg-Vos, L. H. Van Den Berg, J. Visser, M. de Visser, H. Franssen and J. H. Wokke, “The Spectrum of Lower Motor Neuron Syndromes,” Journal of Neurology, Vol. 250, No. 11, 2003, pp. 1279-1292. doi:10.1007/s00415-003-0235-9
- R. M. van den Berg-Vos, J. Visser, H. Franssen, M. de Visser, J. M. B. V. de Jong, S. Kalmijn, J. H. J. Wokke and L. H. van den Berg, “Sporadic Lower Motor Neuron Disease with Adult Onset: Classification of Subtypes,” Brain, Vol. 126, No. 5, 2003, pp. 1036-1047. doi:10.1093/brain/awg117
- J. C. Meadows, C. D. Marsden and D. G. F. Harriman, “Chronic Spinal Muscular Atrophy in Adults: Part 2. Other Forms,” Journal of Neurological Science, Vol. 9, No. 3, 1969, pp. 551-566. doi:10.1016/0022-510X(69)90094-X
- A. E. Harding, P. G. Bradbury and N. M. Murray, “Chronic Asymmetrical Spinal Muscular Atrophy,” Journal of Neurological Science, Vol. 59, No. 1, 1983, pp. 69-83. doi:10.1016/0022-510X(83)90082-5
- A. Pestronk, V. Chaudhry, E. L. Feldman, J. W. Griffin, D. R. Cornblath, E. H. Denys, M. Glasberg, R. W. Kuncl, R. K. Olney and W. C. Yee, “Lower Motor Neuron Syndromes Defined by Patterns of Weakness, Nerve Conduction Abnormalities, and High Titers of Antiglycolipid Antibodies,” Annals of Neurology, Vol. 27, No. 3, 1990, pp. 316-326. doi:10.1002/ana.410270314
- A. Pestronk, D. R. Cornblath, A. A. Ilyas, H. Baba, R. H. Quarles, J. W. Griffin, K. Alderson and R. N. Adams, “A Treatable Multifocal Motor Neuropathy with Antibodies to GM1 Ganglioside,” Annals of Neurology, Vol. 24, No. 1, 1988, pp. 73-78. doi:10.1002/ana.410240113
- V. Chaudhry, A. M. Corse, D. R. Cornblath, R. W. Kuncl, M. L. Freimer and J. W. Griffin, “Multifocal Motor Neuropathy: Electrodiagnostic Features,” Muscle Nerve, Vol. 17, No. 2, 1994, pp. 198-205. doi:10.1002/mus.880170211
- A. Pestronk, G. Lopate, A. J. Kornberg, J. L. Elliott, G. Blume, W. C. Yee and L. T. Goodnough, “Distal Lower Motor Neuron Syndrome with High-Titer Serum IgM Anti-GM1 Antibodies: Improvement Following Immunotherapy with Monthly Plasma Exchange and Intravenous Cyclophosphamide,” Neurology, Vol. 44, No. 11, 1994, pp. 2027-2031.
- C. M. Ellis, S. Leary, J. Payan, C. Shaw, M. Hu, M. O’Brien and P. N. Leigh, “Use of Human Intravenous Immunoglobulin in Lower Motor Neuron Syndromes,” Journal of Neurolgy, Neurosurgery and Psychiatry, Vol. 67, No. 1, 1999, pp. 15-19. doi:10.1136/jnnp.67.1.15
- A. S. Pakiam and G. J. Parry, “Multifocal Motor Neuropathy without Overt Conduction Block,” Muscle Nerve, Vol. 21, No. 2, 1998, pp. 243-245. doi:10.1002/(SICI)1097-4598(199802)21:2<243::AID-MUS14>3.0.CO;2-2
- K. C. Gorson, A. H. Ropper, L. S. Adelman, E. M. raynor and C. B. Saper, “Chronic Motor Axonal Neuropathy: Pathological Evidence of Inflammatory Polyradiculoneuropathy,” Muscle Nerve, Vol. 22, No. 2, 1999, pp. 266-270. doi:10.1002/(SICI)1097-4598(199902)22:2<266::AID-MUS17>3.0.CO;2-V
- R. Kaji, S. Kusunoki, K. Mizutani, N. Oka, Y. Kojima, N. Kohara and J. Kimura, “Chronic Motor Axonal Neuropathy Associated with Antibodies Monospecific for N-acetylgalactosaminyl GD1a,” Muscle Nerve, Vol. 23, No. 5, 2000, pp. 702-706. doi:10.1002/(SICI)1097-4598(200005)23:5<702::AID-MUS6>3.0.CO;2-A
- J. S. Katz, R. J. Barohn, S. Kojan, G. I. Wolfe, S. P. Nations, D. S. Saperstein and A. A. Amato, “Axonal Multifocal Motor Neuropathy without Conduction Block or Other Features of Demyelination,” Neurology, Vol. 58, No. 4, 2002, pp. 615-620.
- J. R. Burrell, C. Yiannikas, D. Rowe and M. C. Kiernan, “Predicting a Positive Response to intravenous Immunoglobulin in Isolated Lower Motor Neuron Syndromes,” PLoS One, Vol. 6, No. 10, 2011, p. e27041. doi:10.1371/journal.pone.0027041
- N. Strigl-Pill, A. König, M. Schröder, H. Beranek, B. G. H. Schoser, M. Spaeth, D. Pongratz and W. Müller-Felber, “Prediction of Response to IVIg Treatment in Patients with Lower Motor Neurone Disorders,” European Journal of Neurology, Vol. 13, No. 2, 2006, pp. 135-140. doi:10.1111/j.1468-1331.2006.01142.x
Abbreviations
IVIg: intravenous immunoglobulin; LMNS: lower motor neuron syndrome; MMN: multifocal motor neuropathy; CIDP: chronic inflammatory demyelinating polyradiculoneuropathy, mMRC: modified Medical Research Council; MRI: magnetic resonance imaging; CMAP: compound muscle action potential; ALP: alkaline phosphatase; NSE: neuron specific enolase; ALS: amyotrophic lateral sclerosis.
NOTES
*Corresponding author.