Open Journal of Modern Neurosurgery
Vol.2 No.3(2012), Article ID:21181,5 pages DOI:10.4236/ojmn.2012.23012
Chordomas and Their Consideration in the Radiographic Differential of Extra-Axial Lesions of the Central Nervous
1Department of Neurosurgery, University of Pennsylvania Medical Center, Philadelphia, USA
2Department of Radiology, Georgetown University Hospital, Washington DC, USA
3Department of Pathology and Laboratory Medicine, Division of Neuropathology, University of Pennsylvania Medical Center, Philadelphia, USA
4Perelman School of Medicine, University of Pennsylvania, Philadelphia, USA
Email: *shihshan.lang@uphs.upenn.edu
Received April 11, 2012; revised May 20, 2012; accepted June 17, 2012
Keywords: Chordoma; Primitive Notochord Tumor; Extra-Axial Tumor; Spine
ABSTRACT
Due to the rare occurrence of chordomas extra-axially, these lesions have not earned a great deal of consideration in the clinical and radiographic differential diagnoses of extra-axial paraspinal lesions. We describe a case of a patient with a surgically resected extra-axial chordoma and review the radiologic characteristics of chordomas as an entity in the spectrum of extra-axial lesions. A 47-year-old man presented to our institution with four months of intermittent parethesias, pain, and subjective weakness in the left leg. MR imaging of the lumbar spine demonstrated a large heterogeneouslyenhancing paraspinal lesion extending from the left L3-4 neural foramen into the psoas muscle. The patient underwent a CT-guided needle biopsy demonstrating features consistent with a chordoma. He then underwent a left retroperitoneal approach and en bloc resection of the lesion from the vertebral column. Pathology confirmed chordoma as the diagnosis. EACs are a rare but important consideration in the diagnosis of extra-axial lesions of the central nervous system.
1. Introduction
Chordomas are histologically benign but locally aggressive tumors which originate from embryonic remnants of the primitive notochord. As such, they arise from persisting elements of the fetal axial skeleton anywhere from Rathke’s pouch to the coccyx. These tumors account for approximately 1% of all intracranial tumors and 4% of primary bone tumors involving the spine [1]. They may occur at any age but are most commonly found in adults ages 30 - 60 and are more common in Caucasians. They do not commonly metastasize (19% of cases [2]) and are frequently diagnosed due to symptoms caused by impingement on adjacent neural structures. Chordomas are not often bounded by capsules, accounting for the high frequency of recurrence following surgical resection [3,4] . Radiation with proton beam or linear accelerator therapy has been advocated to complement neurosurgical therapy [5]. Given the diversity of therapeutic and prognostic factors associated with extra-axial lesions of the spine, the aim of this case report is to discuss the radiological features of chordoma versus a spectrum of other lesions in the differential diagnosis of extra-axial lesions.
2. Case Report
A 47-year-old man presented to our clinic with four months of intermittent parethesias, sharp, electrical pain, and subjective weakness affecting his left buttock radiating down the back of his left leg. These symptoms were most prominent when sleeping on his left side. He was full strength (5/5) in all extremities. He had no other significant medical or family history.
MR imaging of the lumbar spine (Figures 1(A), (B) and (D)) demonstrated a 7 cm heterogeneously-enhanceing paraspinal lesion extending from the left lumbar L3-4 neural foramen into the left psoas muscle, without definite muscular invasion by imaging. The mass was relatively well defined and macrolobulated in contour, with marked T2 hyperintensity (Figures 1(C) and (E)). Multiple internal enhancing septations were also present. Focal signal abnormality and enhancement were also seen in the posterolateral inferior L3 vertebral body, just anterior to the left pedicle, perhaps representing focal osseous involvement. The exiting left L3 nerve root could not be definitively identified. No edematous change was seen in the adjacent left psoas muscle on MRI, suggestive of a slow growing process.
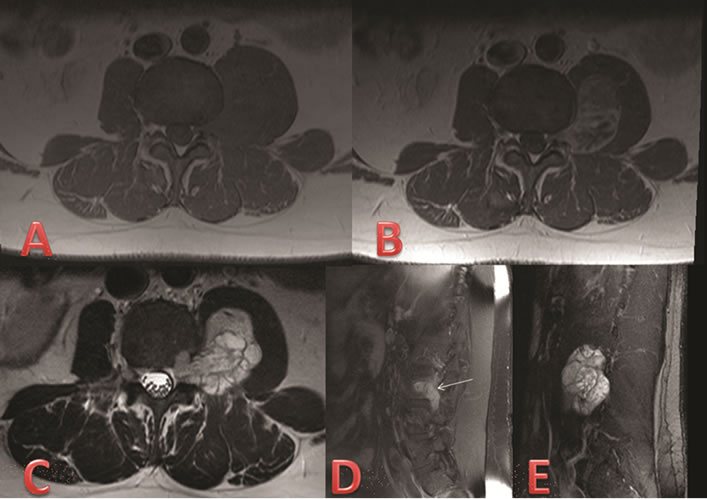
Figure 1. Pre-operative MRI. (A) Axial T1 MR sequence through the L3-4 level demonstrates an isointense left paraspinal mass; (B) Axial T1 post-gadolinium MR sequence shows heterogeneous contrast enhancement of the left paraspinal mass; (C) Axial T2 MR sequence through the L3-4 level demonstrates a hyperintense multi-lobulated left paraspinal mass with internal septations; (D) Sagittal T1 postgadolinium MR sequence depicts the mass extending from the left L3-4 neural foramen into the left psoas muscle. Arrow indicates area of potential vertebral body involvement; (E) Sagittal T2 MR sequence shows extension over L3-4 vertebral levels.
After consultation with a neurosurgeon, the patient was referred to obtain a tissue diagnosis by a CT-guided needle biopsy. After histopathological stains confirmed tissue diagnosis of a chordoma, a definitive open surgical operation to remove the tumor was recommended. In the operating room, the patient was positioned for a left-sided retroperitoneal approach. Dissection through the left-sided retroperitoneal incision was carried out to expose the left aspect of the lumbar vertebral column. The psoas major was divided to expose the paraspinal mass and facilitate exposure of the L2, L3, and L4 nerve roots. Grossly, the mass was firm, immobile, and adherent to the aforementioned nerve roots. Following stimulation of the nerve roots with neuromonitoring, dissection was carried out carefully in an inferior to superior manner using blunt dissection and Metzenbaum scissors. The tumor separated from its surrounding tissue and was removed en bloc with muscular attachments addressed with bipolar cauterization. Gross total resection was performed except for a small component in the foramen that was not well visualized intraoperatively. Post-operative MRI demonstrated a small amount of residual enhancement in the left L3-4 foramen best seen on the sagittal sequence (Figures 2(A) and (B)).
Postoperatively, the patient was full strength on exam (5/5) with improved preoperative paresthesias and pain. On the six week follow-up MRI, this lesion enlarged in size along the left L3-4 foramen (Figures 2(C) and (D)). After recommendation for adjuvant therapy from radiation oncology, he was referred for proton beam therapy.
Histologic examination showed a neoplasm composed of epithelioid cells with eosinophilic bubbly cytoplasm, including cells with signet ring morphology (Figures 3(A) and (B)). Tumor cells displayed mild to moderate atypia and were arranged in nests, cords and sheets in an amorphous mucoid matrix. No significant mitotic activity or necrosis was seen. Immunohistochemical stains showed expression of cytokeratin AE1/AE3 and S-100 protein (Figures 3(C) and (D)). These findings established the diagnosis of chordoma.
3. Discussion
The identification of an extra-axial paraspinal lesion impinging on neural elements commonly raises concern for the presence of metastatic disease, schwannoma, or neurofibroma. However, the potential presence of chondrosarcoma, giant cell tumor, and other entities (including chordoma) must be considered. The pathological basis of how extra-axial chordomas develop is unclear. They are believed to arise from migrating or ectopic notochord, but have been documented arising from distal bones and
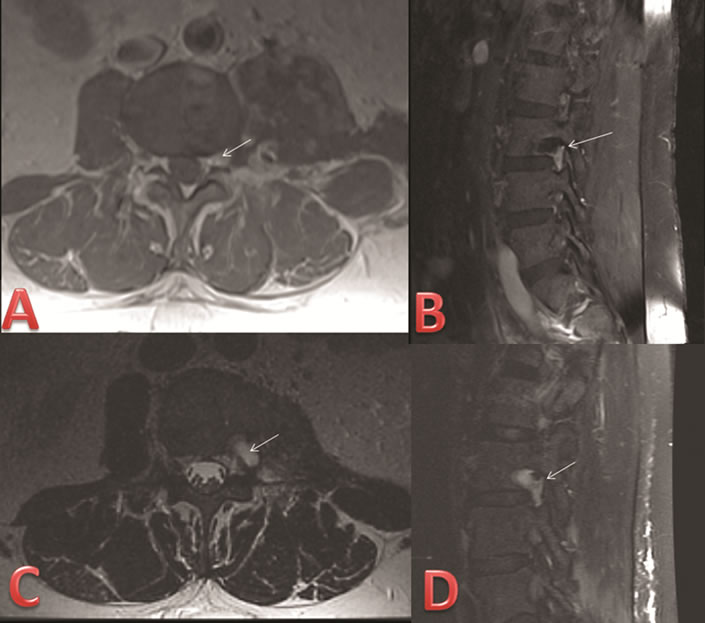
Figure 2. Post-operative MRI. (A) Axial T1 post gadolinium MR sequence through the L3-4 level on postoperative day #1 demonstrates a small focus of enhancing tissue at the left neural foramen suggestive for mild tumor residual (arrow); (B) Sagittal T1 post gadolinium MR sequence through the left L3-4 foramen postoperative day #1 shows similar enhancing focus likely representing mild residual disease (arrow); (C) Axial T2 MR sequence through L3-4 six weeks after the operation demonstrates mild increase in T2 hyperintense material at the operative site (arrow), portions of which likely represent postoperative fluid; (D) Sagittal T1 post-gadolinium MR sequence through the left L3-4 foramen six weeks after the operation demonstrates mildly more prominent tissue enhancement suspicious for residual/recurrent disease (arrow).
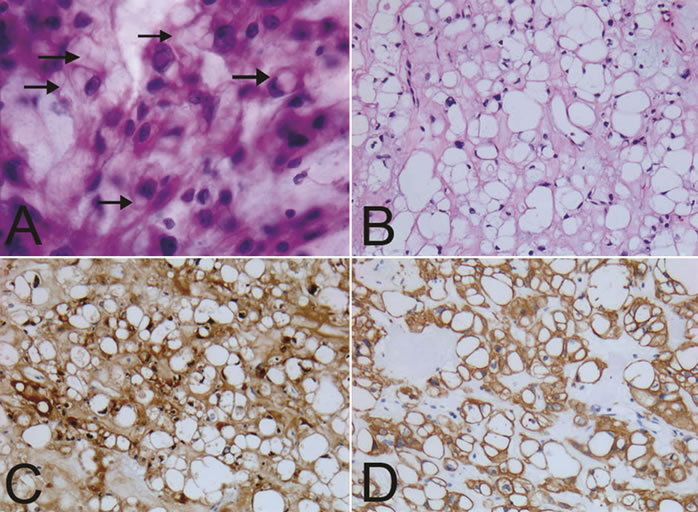
Figure 3. Histolopathology. (A) Smear preparation showing epithelioid tumor cells with eosinophilic to bubbly cytoplasm (arrows) embedded in an amorphous mucoid matrix. Nuclear atypia was mild to moderate. H & E. 40×; (B) Formalin fixed paraffin embedded sections showing epithelioid tumor cells in a mucoid matrix. H & E. 20×; (C) Cytokeratin AE1/AE3 was present in virtually all cells. Cytokeratin AE1/3. 20×; (D) Tumor cells were also positive for S100. 20×.
soft tissue, including lung parenchyma [6-8] . A summary of radiological characteristics of tumors included in the differential diagnosis is highlighted in Table 1.
3.1. Schwannomas and Neurofibromas
Schwannomas have been known to cause several bony changes, including pedicle erosion, neural foraminal widening, and vertebral body scalloping. They may be associated with hemorrhage, cysts, or fatty degeneration. MR imaging often demonstrates a well defined, solitary lesion with T2 hyperintensity and homogeneous enhancement. Surgical excision is considered curative (except with schwannomas in neurofibromatosis), however, structural changes caused by tumor growth may result in spinal instability following excision.
Neurofibromas can have similar imaging characteristics to schwannomas, although neurofibromas are often multiple, typically do not show hemorrhage, cysts, or fatty change. Neurofibromas also more often show a “target” sign on MRI in which there is peripheral enhancement and central hypoenhancement. Multinodular, often infiltrative lesions are termed plexiform neurofibromas and are associated with neurofibromatosis. The vast majority of spinal nerve root tumors in patients with NF-1 are neurofibromas, although both neurofibromas and schwannomas may occur in the absence of NF-1. Calcification is not commonly associated with neurofibromas. Surgical excision is considered more difficult as neurofibromas typically have greater fascicular involvement and tend to be invested within the nerve body. Solitary lesions of the spine occur in the ratio of 3:1 in favor of schwannomas.
3.2. Metastases
Metastastic tumors comprise the vast majority of extra-axial tumors, with the largest part being osteolytic metastases. Most metastases are extradural, while less than 5% are intradural and intramedullary [9]. Lymphoma, lung, breast, and prostate cancer represent the most common subset of osteolytic lesions, though approximately 10% of all patients with cancer will have spinal epidural metastases [1]. Patient’s history of cancer will help to direct diagnosis. Radiographically, these lesions may differ based on their respective cellular origin. Typically metastases are T1 hypointense, T2 hyperintense, with enhancement, although with an increasing sclerotic component there may be decreased T2 hyperintensity and enhancement. Treatment is palliative in nature, consisting of either surgery alone or surgery plus radiation therapy and many are also treated with radiation alone.
3.3. Chondrosarcomas
Chondrosarcomas are associated with bony destruction and extension into nearby soft tissues. CT or plain film imaging may best detect chondroid calcification, often seen in the soft tissue component of the mass, and is the radiographic hallmark of this entity. Chondrosarcomas are uncommonly seen in the spine, but when present are often thoracic in location. They can involve the posterior elements, vertebral corpus, or both. Wide surgical resection is advised, with post-operative radiation if full resection cannot be accomplished [10].
3.4. Giant Cell Tumors
Giant cell tumors (GCTs) are uncommonly seen in the spine, with sacral lesions accounting for the largest subset of spinal lesions. Radiographically, GCTs are characterized as lytic, expansile lesions with eccentric bone destruction on plain films and CT imaging, exhibiting “a soap bubble effect”. These masses are often very heterogeneous on both CT and MR imaging and frequently demonstrate cystic change, internal hemorrhage, and fluid-fluid levels. Giant cell tumors are vascular masses and show enhancement on post-contrast imaging. Marginal excision with or without pre-operative embolization is advocated, though complete sacrectomy may be too extreme when cement implantation can control the lesion [11].
3.5. Chordomas
Chordomas are malignant tumors which arise from notochord remnants, and therefore are typically midline or central in location anywhere from the Rathke’s pouch to the coccyx. Lesions are most commonly sacrococcygeal, with decreasing frequency in the spheno-occipital region,
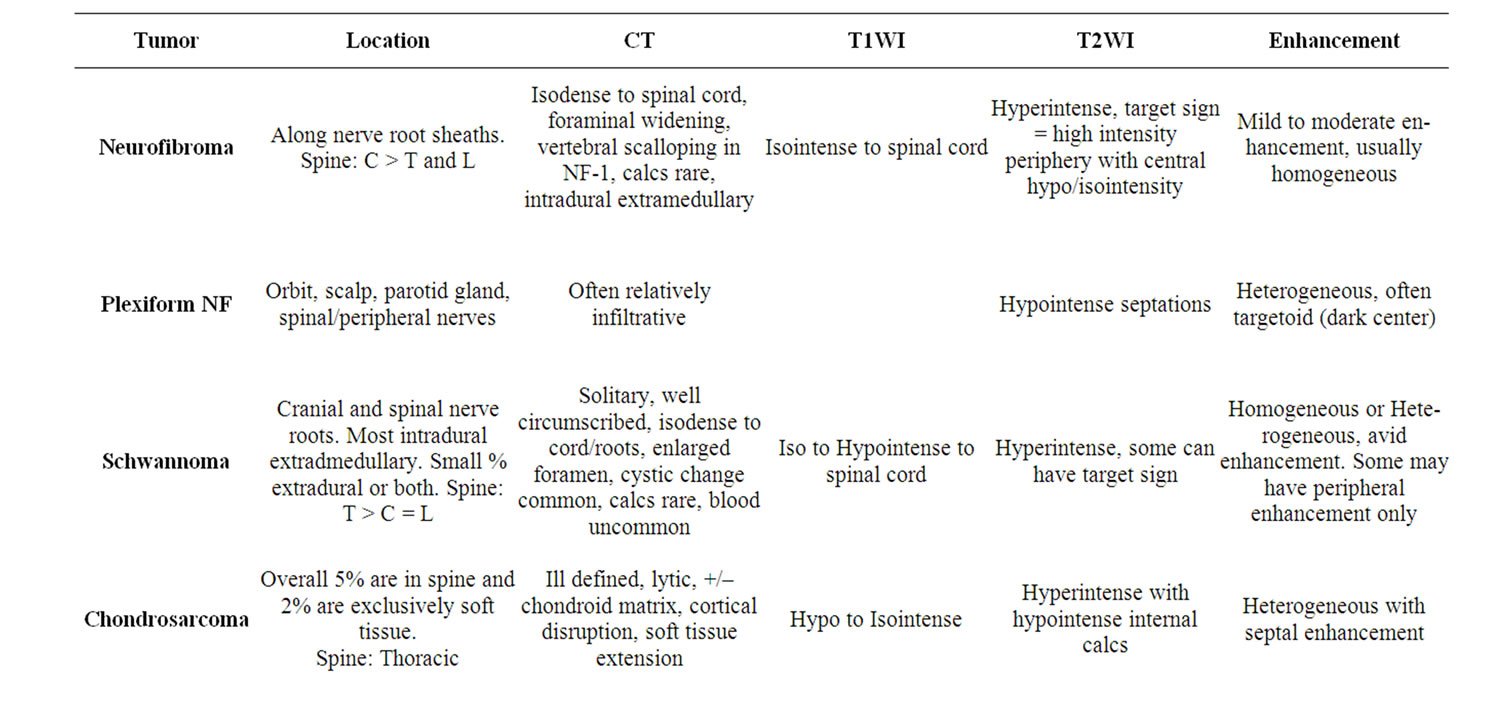

Table 1. Summary of radiologic appearance of tumors included in the differential diagnosis with chordoma.
and vertebral body axis. Clival chordomas typically project in the midline and impinge upon the pons, producing a characteristic “thumb sign”. Chordomas of the vertebral bodies usually affect cervical and lumbar segments. Vertebral body lesions are often lytic bone lesions, at times with a sclerotic rim and calcification. Vertebral lesions may involve the disc space and contiguous vertebrae. CT imaging demonstrates a soft-tissue like mass with bony destruction and areas of calcification. On MR imaging, lesions appear hyperintense on T2WI with avid enhancement. On T1WI, chordomas are isointense to the spinal cord although areas of hypointense calcification may be identified. Chordomas are generally not sensitive to radiation or chemotherapy, consequently en bloc resection is advised. Intensity modulated radiation therapy (IMRT) and stereotactic radiosurgery (i.e. CyberKnife® or Gamma Knife®) are sometimes used to treat chordomas with varying results and relatively high complication rates in the cranial base location [12,13] . Chordomas are generally resistant to standard cytotoxic chemotherapy agents, however, some success has been reported with Gleevec® (imatinib) [14] and epidermal growth factor receptor inhibitors such as cetixumab and gefitinib [15,16] . Advances in photon and proton radiation therapy may provide positive results for poor surgical candidates and patients with recurrent tumor [17].
4. Conclusion
EACs are a rare but important consideration in the diagnosis of extra-axial lesions of the central nervous system. This case report emphasizes the need for neurosurgeons to be familiar with the variations in MRI appearance among these different entities because of its implications in presurgical planning. Due to the finding that conventional radiation therapy and chemotherapy are generally not useful treatments in chordomas, it is recommended that these patients undergo an aggressive surgical approach with the goal of a gross total resection. In these large paraspinous lesions that could represent a broad range of diagnoses, diagnostic needle biopsy is of great value as the results can effectively guide surgical planning. Survival, recurrence rates and rates of metastasis are largely unknown because of the rarity of this condition. More research is needed to define the role of photon therapy and newer advances in radiation and chemotherapy for those patients in which gross total resection cannot be achieved.
REFERENCES
- K. D. Barron, A. Hirano, S. Araki and R. D. Terry, “Experiences with Metastatic Neoplasms Involving the Spinal Cord,” Neurology, Vol. 79, No. 2, 1959, pp. 91-106. doi:10.1212/WNL.9.2.91
- C. M. McPherson, D. Suki, I. E. McCutcheon, Z. L. Gokaslan, L. D. Rhines and E. Mendel, “Metastatic Disease from Spinal Chordoma: A 10-Year Experience,” Journal of Neurosurgery: Spine, Vol. 5, No. 4, 2006, pp. 277-280. doi:10.3171/spi.2006.5.4.277
- S. A. Hanna, W. J. Aston, T. W. Briggs, S. R. Cannon and A. Saifuddin, “Sacral Chordoma: Can Local Recurrence after Sacrectomy Be Predicted?” Clinical Orthopaedics and Related Research, Vol. 466, No. 9, 2008, pp. 2217-2223. doi:10.1007/s11999-008-0356-7
- J. E. York, A. Kaczaraj, D. Abi-Said, G. N. Fuller, J. M. Skibber, N. A. Janjan, et al., “Sacral Chordoma: 40-Year Experience at a Major Cancer Center,” Neurosurgery, Vol. 44, No. 1, 1999, pp. 74-80. doi:10.1007/s11999-008-0356-7
- A. M. Berson, J. R. Castro, P. Petti, T. L. Phillips, G. E. Gauger, P. Gutin, et al., “Charged Particle Irradiation of Chordoma and Chondrosarcoma of the Base of Skull and Cervical Spine: The Lawrence Berkeley Laboratory Experience,” International Journal of Radiation Oncology & Biology & Physics, Vol. 15, No. 3, 1988, pp. 559-565. doi:10.1016/0360-3016(88)90295-7
- P. O’Donnell, R. Tirabosco, S. Vujovic, W. Bartlett, T. W. Briggs, S. Henderson, et al., “Diagnosing an Extra-Axial Chordoma of the Proximal Tibia with the Help of Brachyury, a Molecule Required for Notochordal Differentiation,” Skeletal Radiology, Vol. 36, No. 1, 2007, pp. 59-65. doi:10.1007/s00256-006-0167-4
- S. Y. Park, S. R. Kim, Y. H. Choe, K. Y. Lee, S. J. Park, H. B. Lee, et al., “Extra-Axial Chordoma Presenting as a Lung Mass,” Respiration, Vol. 2, 2009, pp. 219-223. doi:10.1159/000134306
- A. C. van Akkooi, A. N. van Geel, J. H. Bessems and M. A. den Bakker, “Extra-Axial Chordoma,” Journal of Bone & Joint Surgery, British Volume, Vol. 88-B, No. 9, 2006, pp. 1232-1234. doi:10.1302/0301-620X.88B9.17873
- U. Schick, G. Marquardt and R. Lorenz, “Intradural and Extradural Spinal Metastases,” Neurosurgical Review, Vol. 24, No. 1, 2001, pp. 1-5. doi:10.1007/PL00011959
- T. C. Shives, R. A. McLeod, K. K. Unni and M. F. Schray, “Chondrosarcoma of the Spine,” Journal of Bone & Joint Surgery, Ameican Volume, Vol. 71, No. 8, 1989, pp. 1158-1165.
- T. Ozaki, U. Liljenqvist, H. Halm, A. Hillmann, G. Gosheger and W. Winkelmann, “Giant Cell Tumor of the Spine,” Clinical Orthopaedics and Related Research, Vol. 401, 2002, pp. 194-201. doi:10.1097/00003086-200208000-00022
- M. A. Torres, E. L. Chang, A. Mahajan, D. G. Lege, B. A. Riley, X. Zhang, et al., “Optimal Treatment Planning for Skull Base Chordoma: Photons, Protons, or a Combination of Both?” International Journal of Radiation Oncology & Biology & Physics, Vol. 74, No. 4, 2009, pp. 1033- 1039. doi:10.1016/j.ijrobp.2008.09.029
- S. Krishnan, R. L. Foote, P. D. Brown, B. E. Pollock, M. J. Link and Y. I. Garces, “Radiosurgery for Cranial Base Chordomas and Chondrosarcomas,” Neurosurgery, Vol. 56, No. 4, 2005, pp. 777-784. doi:10.1227/01.NEU.0000156789.10394.F5
- P. G. Casali, A. Messina, S. Stacchiotti, E. Tamborini, F. Crippa, A. Gronchi, et al., “Imatinib Mesylate in Chordoma,” Cancer, Vol. 101, No. 9, 2004, pp. 2086-2097. doi:10.1002/cncr.20618
- H. Hof and T. Welzel, J. Debus, “Effectiveness of Cetuximab/Gefitinib in the Therapy of a Sacral Chordoma,” Onkologie, Vol. 29, No. 12, 2006, pp. 572-574.
- S. G. Launay, B. Chetaille, F. Medina, D. Perrot, S. Nazarian, J. Guiramand, et al., “Efficacy of Epidermal Growth Factor Receptor Targeting in Advanced Chordoma: Case Report and Literature Review,” BMC Cancer, Vol. 11, 2011, p. 423. doi:10.1186/1471-2407-11-423
- D. M. Sciubba, J. H. Chi, L. D. Rhines, Z. L. Gokaslan, “Chordoma of the Spinal Column,” Neurosurgery Clinics of North America, Vol. 19, No. 1, 2008, pp. 5-15. doi:10.1016/j.nec.2007.09.006
NOTES
*Corresponding author.