Open Journal of Endocrine and Metabolic Diseases
Vol. 3 No. 1 (2013) , Article ID: 28029 , 4 pages DOI:10.4236/ojemd.2013.31006
Functioning Adrenocortical Oncocytic Carsinoma: A Case Report
1Department of Endocrinology and Metabolism, Faculty of Medicine, Yuzuncu Yil University, Van, Turkey
2Department of Internal Medicine, Van Education and Research Hospital, Van, Turkey
3Department of Endocrinology and Metabolism, Faculty of Medicine, Medipol University, İstanbul, Turkey
Email: drmuratatmaca@hotmail.com
Received November 12, 2012; revised December 15, 2012; accepted January 16, 2013
Keywords: Oncocytoma; Cortisol; Testosterone
ABSTRACT
Introduction: Oncocytic neoplasms of the adrenal cortex are extremely rare and are usually nonfunctioning. Only a little more than fifty cases have been described in the literature. Case Presentation: We report the case of an adrenocortical oncocytic neoplasm with malignant potential in a 21-year-old female with co-secrete testosterone and cortisol. The patient had been operated of adrenal mass. Fallowing operation, the Cushing’s syndrome and high testosterone levels resolved. Conclusion: In our knowledge this is the first case in the lıterature which co-secret cortizol and testosterone in malign oncystoma.
1. Introduction
The term “oncocyte”, first used by Hamperl in 1950, describes large, highly eosinophilic, granular cells associated with a hurthle cell tumor of the thyroid gland [1]. An oncocytoma is a tumor which is composed predominantly of polygonal oncocytes with abundant granular and intensely eosinophilic cytoplasm [2]. It occurs in several organs, most commonly in the kidney, salivary glands, and thyroid and less commonly in lung, parathyroid gland, ovary or pituitary gland [2]. Adrenocortical oncocytoma is very rare. The first case was reported in 1986, and only a little more than fifty cases have been described in the literature [3,4]. Adrenocortical oncocytic neoplasms represent unusual lesions and three histological categories are included: oncocytoma, oncocytic neoplasm of uncertain malignant potential and oncocytic carcinoma [5]. Most of these adrenal oncosytic neoplasms are detected incidentally and usually benign and nonfunctioning [6]. We report here a rare case of functioning adrenocortical oncocytic carsinoma which cosecrete testosterone and cortisol.
2. Case Report
A 21-year-old woman was admitted to our hospital with complaints of amenorrhea, hirsutism, hair drop and hoarseness. Her menarche age was 13. Her menstruation periods were regular for the first five years but for the last three years she has not had menstruation cycle. Our patient who has been married for two years has been treated with oral contraceptive pills because of amenorrhea as a result she had menstruation cycle several months. Her hirsutism and hoarseness complaints started two years ago and increased gradually. There is no medical specialty neither in her nor in her family history. In the physical examination her arterial blood pressure was 90/60 mmHg. There was no cushinglike appearance but there was hairing on her chin, breast and lower abdomen regions. Except these there was not any other specialty in physical examinations. In laboratory examination leucocytosis and polycythemia have been detected. The glucose and electrolyte levels were normal. The hormonal surveys to evaluate hirsutism and amenorrhea are given in Table 1. Cortisol was not suppressed in overnight, low dose and high dose dexamethasone suppression tests. Testosterone level was very high and ACTH level was supresed. As a result we suspected cortisol and testosterone secreting adrenal cancer.
After examining the abdominal computerized tomography in left adrenal gland, 5.3 cm diameter well circumscribed hyper dense tumoral lesion was detected (Figure 1). Laparoscopic adrenalectomy was performed with steroid protection. Histopathological examination revealed a well circumscribed, solid tumour, which mea
Table 1. Laboratuary results of patients.
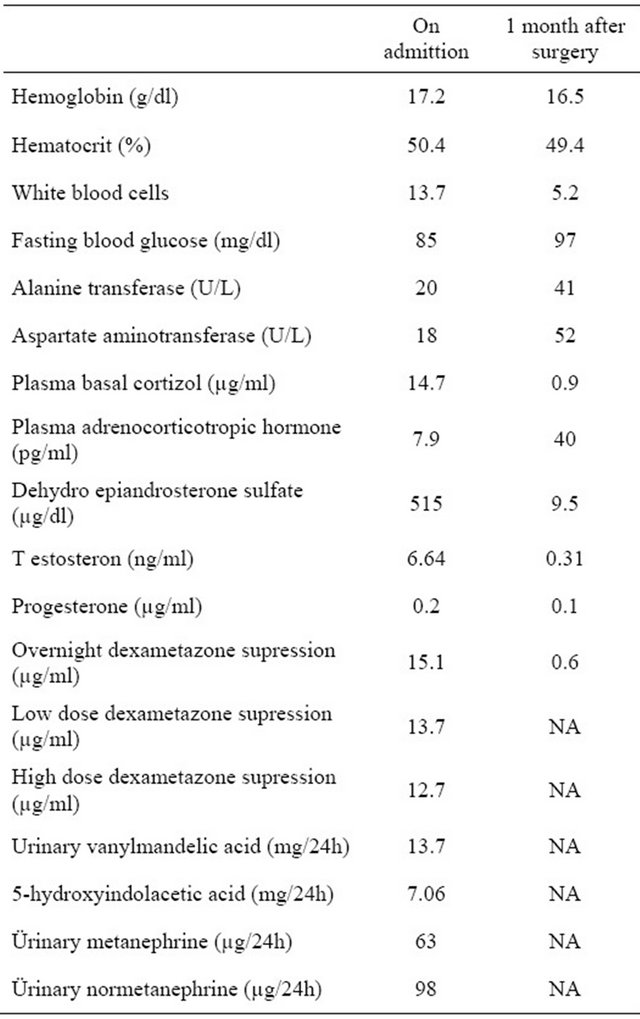
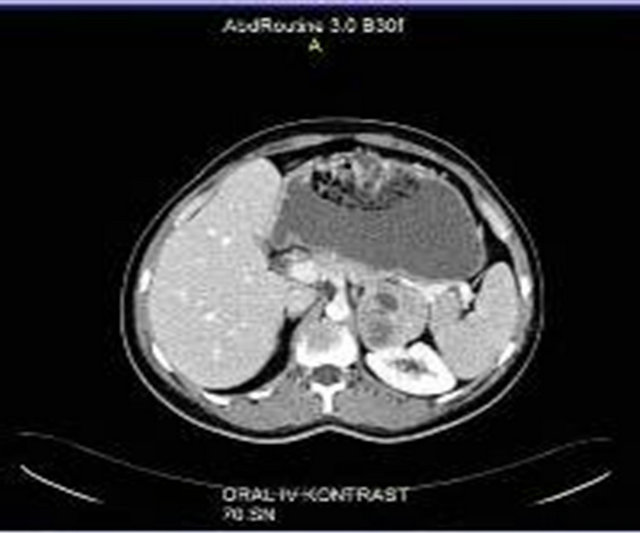
Figure 1. Well circumscribed hyperdense tumoral lesion was shown in left adrenal gland.
sured 5.5 × 4.5 × 4 cm. On the periphery of the tumor, residual adrenal tissue was seen focally. The cut surface was solid and yellow-beige. There were no cystic degenerations or necrosis, but there were haemorrhages focallly. Microscopically, the tumour was surround thin a capsule and composed of moderate to large polygonal cells with abundant, eosinophilic and granular cytoplasm (Figure 2). The nuclei were round or oval with a single, large and prominent nucleolus. Bizarre, highly pleomorphic nuclei (nuclear atypia) and nuclear pseudoinclusions were also seen focally. There was no evidence of necrosis, capsular or sinusoidal invasion, but there was lymphovascular invasion on the periphery of the tumor (Figure 3). Mitotic figures were identified, numbering 1 per 50 high power fields. No atypical mitotic figures were found. The proliferative index Ki-67 was less than 1%. Immunohistochemically, tumor cells ChromograninA, Pancytokeratin and NSE were negative. Pathologic diagnose was oncocytoma and because of peritumoral invasion it was anticipated that it will behave malign. After surgery, the Cushing’s syndrome and high testosterone levels resolved (Table 1). The case became pregnant two months after operation. After a healthy pregnancy she had a son. The patient is still alive with no metastases, 24 months after the surgery.
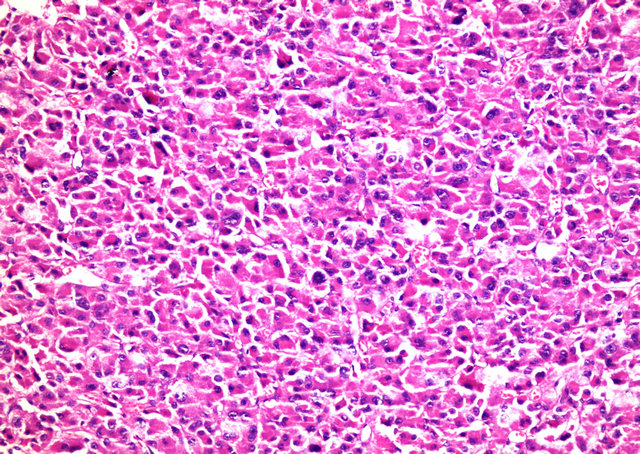
Figure 2. Adrenocortical oncocytoma. Tumor cells contain densely granular eosinophilic cytoplasm and pleomorphic nuclei (H-E ×200).
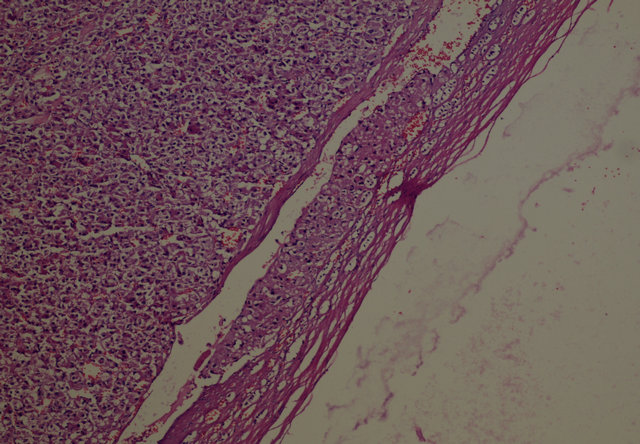
Figure 3. Lymphovascular invasion on the periphery of the tumor (H-E ×100).
3. Discussion
Most of the adrenocortical oncocytomas were well circumscribed, ranging from 2.2 to 15 cm (mean 8 cm) and a mean weight 281 g. The majority arises in females between 40 and 50 years of age (mean 47.6) [7,8]. In our case the patient is female and below the average age that is 21. The masses tend to be brown, yellow or mahogany on cut sections. Some tumours show areas of haemorrhage and necrosis. The classic central radiating scar that has been described in renal oncocytomas is not always present in adrenocortical oncocytomas. The microscopic appearance of oncocytomas includes cells arranged in solid, trabecular, tubular or papillary patterns. The tumor cells are highly eosinophilic and granular, which may be attributed to the presence of numerous mitochondria as seen by electron microscopy. The neoplasm cells seldom have pleomorphic nuclei or mitotic figure [9,10]. In our case in addition to the highly eosinophilic and granular tumor cells, necrosis was negative, pleomorphism and atypia were significant. The role of the proliferative index (Ki-67) and oncoprotein p53 has also been a controversial issue in the past years. Some authors have suggested that these markers could be used as potential indicators of the benign or malignant nature of adrenocortical carcinomas [11-13]. In our case pathologic diagnose was oncocytoma and because of peritumoral invasion it was anticipated that it will behave malign. Ki-67 proliferation index of cells was less than 1%. Bisceglia et al. results concerning Ki-67 expression of Adrenocortical oncocytic neoplasms were mostly in accordance with previous studies of the proliferative index in conventional adrenocortical carcinomas [14]. However, other authors’ studies showed that Ki-67 as long as p53 cannot be reliably used to predict the biological behaviour of Adrenocortical oncocytic neoplasms [6,15,16]. Generally, oncocytoma is regarded as a benign tumour, but a malignnant variant with local invasion and distant metastases was described in 1991; it had invaded the inferior vena cava and metastasized to the liver [17]. In our case the capsule invasion was negative, vessel invasion was positive in peritumoral region. Kurek et al. described a patient who exhibited widespread retroperitoneal infiltration 7 years after the removal of an adrenal tumor [18]. Malignant functioning oncocytic tumor in literature was only reported in 2007. In that case cortisol secreted tumor infiltrated to the vena cava inferior [19]. It was detected that the malignant functioning oncocytic tumor was secreting cortisol and testosterone in our case.
The biologic behavior of adrenocortical neoplasms is the most important practical problem. A combination of clinical, biochemical and histological features can differentiate benign and malignant adrenocortical tumors. Aside from the obviously malignant cases in which the diagnosis is based on common clinicopathologic features universally valid for tumors of any site, such as distant metastases, surgical unresectability and/or invasion of adjacent organs, the prediction of biologic behavior and clinical outcome can be difficult. There is no single histological parameter that is predictive of the clinical outcome and biologic behavior of adrenocortical oncocytic neoplasms. The system of Weiss, which is most commonly used, has been developed for distinguishing benign from malignant adrenocortical neoplasms [20]. Recently Bisceglia et al. proposed new criteria with a modified form to classify the adrenocortical oncocytic tumors. If the oncocytic tumor exhibits one or more of the major criteria (a mitotic rate of more than 5 mitoses per 50 high power fields, any atypical mitosis or any venous invasion), it is considered to be malignant. If the tumor exhibits one or more of the minor criteria [large sized tumor (>10 cm and/or >200 g), necrosis, capsular invasion, or sinusoidal invasion], the tumor is considered to be borderline (uncertain malignant potential). If none of the aforementioned features are present, the tumor can be considered benign [14]. In our case pathologic diagnose was oncocytoma and because of peritumoral invasion it was anticipated that it will behave malign.
Computed tomography and magnetic resonance images have no special appearances for adrenal oncosytoma [21]. Their MRI appearance is not distinguishable from malignancy, and the stellate scar seen in larger renal oncocytomas has not been reported in the adrenal [21]. Adrenocortical oncocytomas can show increased 18FFDG uptake, the cause may be the intense hypermetabolism of glucose and the presence of numerous intracellular mitochondria [22].
There is a wide discussion about the multimodality therapeutic approach which is needed apart from the radical surgical excision of the primary adrenocortical carcinoma tumour, its local recurrences and relevant metastatic involved sites [23,24]. We performed left adrenalectomy in our case.
Radiotherapy, in adjuvant or symptomatic control setting seems to be delivered helpfully or as a standard care of palliation [23]. In clinical trials, metastatic adrenocortical carcinoma extensive disease is treated with mitotane and multiple chemotherapeutic regiments combination (i.e. etoposide, doxorubicin, cisplatin or streptozotocin). Chemotherapy in adjuvant setting is under discussion so far [24]. The mainstay therapeutic approach in both adrenocortical carcinomas and oncocytic carcinomas is wide surgical resection. In oncocytic carcinomas, radiotherapy, mitotane and/or chemotherapy is given individually post bulky cytomassive excision, depending on disease staging and predominant symptoms.
REFERENCES
- H. Hamperl, “Onkocytes and the so Called Hurthle Cell Tumor,” Archives of Pathology, Vol. 49, 1950, pp. 563-570.
- A. Chang and S. J. Harawi, “Oncocytes, Oncytosis and Oncytotic Tumors,” Pathology Annual, Vol. 27, No. 1, 1992, pp. 263-304.
- S. Kakimoto, Y. Yushita, T. Sanefugi, A. Kondo, N. Fujishima, M. Kishikawa, et al., “Non-Hormonal Adrenocortical Ademona with Oncocytoma Like Appearances,” Hinyokika Kiyo. Acta Urologica Japonica, Vol. 32, No. 5, 1986, pp. 757-763.
- X. J. Tian,Y. Huang, J. Lu and L. L. Ma, “Laparoscopic Adrenocortical Oncocytoma Resection,” Chinese Medical Journal, Vol. 121, No. 9, 2008, pp. 862-864.
- Z. W. Baloch and V. A. LiVolsi, “Oncocytic Lesions of the Neuroendocrine System,” Seminars in Diagnostic Pathology, Vol. 16, No. 2, 1999, pp. 190-199.
- B. T. Lin, S. M. Bonsib, G. W. Mierau, L. M. Weiss and L. J. Medeiros, “Oncocytic Adrenal Neoplasms: A Report of Seven Cases and Review of the Literature,” The American Journal of Surgical Pathology, Vol. 22, No. 5, 1998, pp. 603-614. doi:10.1097/00000478-199805000-00012
- D. Botsios, K. Blouhos, K. Vasiliadis, A. Asimaki, K. Tsalis and D. Betsis, “Adrenocortical Oncocytoma: A Rare Tumor of Undefined Malignant Potential,” Surgery Today, Vol. 37, No. 7, 2007, pp. 612-617. doi:10.1007/s00595-006-3458-4
- G. Mikuz, “Clinical Pathology of Urologic Tumors,” CRC Press, Boca Raton, 2007.
- M. N. Morra and S. Das, “Renal Oncocytoma: A Review of Histogenesis, Histopathology, Diagnosis and Treatment,” Journal of Urology, Vol. 150, No. 2, 1993, pp. 295- 302.
- G. K. Nguyen, R. Vriend, D. Ronagham and W. H. Lakey, “Heterotopic Adrenocortical Oncocytoma: A Case Report with Light and Electron Microscopic Studies,” Cancer, Vol. 70, No. 11, 1992, pp. 2681-2684. doi:10.1002/1097-0142(19921201)70:11<2681::AID-CNCR2820701119>3.0.CO;2-H
- J. R. Goldblum, R. Shannon, E. P. Kaldjian, M. Thiny, R. Davenport, N. Thompson and R. V. Lloyd, “Immunohistochemical Assessment of Proliferative Activity in Adrenocortical Neoplasms,” Modern Pathology, Vol. 6, No. 6, 1993, pp. 663-668.
- M. P. Vargas, H. I. Vargas, D. E. Kleiner and M. J. Merino, “Adrenocortical Neoplasms: Role of Prognostic Markers MIB-1, P53 and RB,” The American Journal of Surgical Pathology, Vol. 21, No. 5, 1997, pp. 556-562. doi:10.1097/00000478-199705000-00008
- J. S. Winston, A. Mompoint, H. Wang and D. Crossland, “The Utility of Ki-67 in the Evaluation of Adrenocortical Neoplasms,” Modern Pathology, Vol. 10, 1997, p. 52A.
- M. Bisceglia, O. Ludovico, A. Di Mattia, D. Ben-Dor, J. Sandbank, G. Pasquinelli, S. K. Lau and L. M. Weiss, “Adrenocortical Oncocytic Tumors: Report of 10 Cases and Review of the Literature,” International Journal of Surgical Pathology, Vol. 12, No. 3, 2004, pp. 231-243. doi:10.1177/106689690401200304
- S. Krishnamurthy, N. G. Ordonez, T. O. Shelton, A. G. Ayala and N. Sneige, “Fine-Needle Aspiration Cytology of a Case of Oncocytic Adrenocortical Carcinoma,” Diagnostic Cytopathology, Vol. 22, No. 5, 2000, pp. 299-303. doi:10.1002/(SICI)1097-0339(200005)22:5<299::AID-DC8>3.0.CO;2-5
- M. P. Hoang, A. G. Ayala and J. Albores-Saavedra, “Oncocytic Adrenocortical Carcinoma: A Morphologic, Immunohistochemical and Ultrastructural Study of Four Cases,” Modern Pathology, Vol. 15, No. 9, 2002, pp. 973- 978. doi:10.1038/modpathol.3880638
- A. K. El-Naggar, D. B. Evans and B. Mackay, “Oncocytic Adrenal Cortical Carcinoma,” Ultrastructural Pathology, Vol. 15, No. 4-5, 1991, pp. 549-556. doi:10.3109/01913129109016262
- R. Kurek, R. Von Knobloch, U. Feek, A. Heidenreich and R. Hofmann, “Local Recurrence of an Oncocytic Adrenocortical Carcinoma with Ovary Metastasis,” Journal of Urology, Vol. 166, No. 3, 2001, p. 985. doi:10.1016/S0022-5347(05)65882-X
- F. Gołkowski, M. Buziak-Bereza, B, Huszno, A. BałdysWaligórska, A. Stefańska, A. Budzyński, K. Okoń, R. Chrzan and A. Urbanik, “The Unique Case of Adrenocortical Malignant and Functioning Oncocytic Tumour,” Experimental and Clinical Endocrinology & Diabetes, Vol. 115 No. 6, 2007 pp. 401-404. doi:10.1055/s-2007-967083
- L. M. Weiss, L. J. Medeiros and A. L. Vickery Jr., “Pathologic Features of Prognostic Significance in Adrenocortical Carcinoma,” The American Journal of Surgical Pathology, Vol. 13, No. 3, 1989, pp. 202-206. doi:10.1097/00000478-198903000-00004
- E. J. Gandras, L. H. Schwartz, D. M. Panicek and G. Levi, “Case Report. Adrenocortical Oncocytoma: CT and MRI Findings,” Journal of Computer Assisted Tomography, Vol. 20, No. 3, 1996, pp. 407-409. doi:10.1097/00004728-199605000-00016
- D. J. Kim, J. J. Chung, Y. H. Ryu, S. W. Hong, J. S. Yu, and J. H. Kim, “Adrenocortical Oncocytoma Displaying Intense Activity on 18F-FDG-PET: A Case Report and a Literature Review,” Annals of Nuclear Medicine, Vol. 22 No. 9, 2008, pp. 821-824. doi:10.1007/s12149-008-0199-z
- B. Allolio and M. Fassnacht, “Clinical Review: Adrenocortical Carcinoma: Clinical Update,” The Journal of Clinical Endocrinology & Metabolism, Vol. 91, No. 6, 2006, pp. 2027-2037. doi:10.1210/jc.2005-2639
- C. I. M. van Ditzhuijsen, R. van de Weijer and H. R. Haak, “Adrenocortical Carcinoma,” Netherlands Journal of Medicine, Vol. 65, No. 2, 2007, pp. 55-60.