Open Journal of Pathology
Vol.3 No.1(2013), Article ID:27295,5 pages DOI:10.4236/ojpathology.2013.31006
Kikuchi-Fujimoto Disease in Patients with Sjögren’s Syndrome
![]()
1College of Dentistry, University of Oklahoma Health Sciences Center, Oklahoma City, USA; 2Arthritis & Clinical Immunology Program, Oklahoma Medical Research Foundation, Oklahoma City, USA.
Email: Lida-radfar@ouhsc.edu
Received September 21st, 2012; revised November 2nd, 2012; accepted November 12th, 2012
Keywords: Sjögren’s Syndrome; Kikuchi-Fujimoto Disease
ABSTRACT
Sjögren’s syndrome is a chronic autoimmune exocrinopathy associated with dry eyes and dry mouth as major clinical manifestations. It is characterized by lymphocytic infiltration of lacrimal and salivary glands and autoantibody production, especially anti-Ro (or SSA) and anti-La (or SSB). Lymphoproliferative disorders are a feature of Sjögren’s syndrome, and can be considered an extraglandular manifestation of the disease. Kikuchi-Fujimoto disease or histiocytic necrotizing lymphadenitis is a rare form of lymphadenitis. It is reported more often in young adult women with localized lymphadenopathy (usually cervical), fever, rashes, and leukopenia. It is a self-limiting disease with resolution within 1 - 4 months in almost all patients. Sjögren’s syndrome has been reported in patients with other systemic diseases including SLE and lymphomas. Here we present a patient with Kikuchi-Fujimoto disease who developed Sjögren’s Syndrome 8 years after her diagnosis of Kikuchi-Fujimoto disease.
1. Introduction
Sjögren’s syndrome is a chronic autoimmune exocrinopathy associated with dry eyes and dry mouth as major clinical manifestations. It is characterized by lymphocytic infiltration of lacrimal and salivary glands and autoantibody production, especially anti-Ro (or SSA) and anti-La (or SSB). The etiology and pathophysiology underlying Sjögren’s syndrome are not clear. Chronic inflammation and persistent salivary gland enlargement in patients with SS are associated with impaired function [1,2].
The clinical features of SS can be divided into exocrine and extraglandular manifestations. All patients with SS have dry mouth and eyes. In a study of 80 patients followed over 7.5 years, Anaya et al. reported dry mouth and eyes in all patients, but only 31% of patients present with only dry mouth and eyes [3]. The same study reported 25% extraglandular involvement in SS. BritoZerón et al. in a study in 2005, reported 20% with circulating monoclonal immunoglobulins in primary SS. In those, monoclonal IgG was the most frequent type of immunoglobulin detected [4].
The lifetime risk of non-Hodgkin’s lymphoma in SS is 5% (16 - 44 times higher than normal population) [5,6]. Transition of SS to lymphoma is a slow process (rate of 6.5 - 7.5 years after diagnosis of SS). Primary SS patients can develop lymphoproliferative disease of the upper respiratory system or enlargement of the lymph nodes of the lung [4]. Thus, lymphoproliferative disorders are a feature of Sjögren’s syndrome, and can be considered an extraglandular manifestation of the disease.
Kikuchi-Fujimoto disease or histiocytic necrotizing lymphadenitis is a rare form of lymphadenitis. It was described in 1972 in Japan by Kikuchi and Fujimoto independently [7,8]. Soon after the first report, cases were reported from other parts of the world [9]. Kikuchi-Fujimoto disease is reported more often in young adult women, with an average age of 30 - 50 years old. The etiology is unknown, but Hudnall et al., 2008 suggested that “the necrotic reaction of late stage Kikuchi-Fujimoto may at least in part be due to apoptosis and phagocytosis of virus-infected lymphoid cells” [10]. Symptoms include localized lymphadenopathy (usually cervical), fever, rashes, and leukopenia. It is a self-limiting disease with resolution within 1 - 4 months in almost all patients [11].
Sjögren’s syndrome has been reported in patients with systemic lupus erythematosus [12-17], and lymphomas. [18] In one series of 61 patients with Kikuchi-Fujimoto disease, 2 went on to develop SLE [19]. In a review of 244 patients with Kikuchi-Fujimoto disease Kucukardali, et al. found 32 (13%) had associated SLE, but no Kikuchi-Fujimoto disease patient had Sjögren’s syndrome [20]. Here we report Kikuchi-Fujimoto disease with Sjögren’s syndrome.
2. Case Report
A 44-year-old white woman presented to a dental office for right facial pain and temporomandibular joint (TMJ) pain of a few months duration. The pain was moderate, occasional and pain medications were helpful. Her past medical history was significant for Kikuchi-Fujimoto disease in 2007. In December 2006 she developed fever, red raised ring on her left forehead and right leg below the knee, cervical lymphadenopathy, arthralgia (bilateral knee pain), and skin rash. She received outpatient antibiotics for several courses and was subsequently admitted to the local hospital to receive IV antibiotics where she developed total body skin rash consisting of red raised, ulcerated lesions. She underwent an excisional biopsy of cervical lymph node which showed necrotizing lymphadenitis (Figures 1(A) and (B)). Thus, the diagnostic work up led to a diagnosis of Kikuchi-Fujimoto disease. Patient underwent a serological investigation that showed a positive ANA at 1:3240 along with positive anti-Ro and anti-La by immunodiffusion. However, there were no other clinical or laboratory features to further suggest either disease. In particular, there were no dry eyes or dry mouth. She was not diagnosed with either SLE or SS, and did not meet the classification criteria for either disease [21,22]. Otherwise, she did not have any other medical problem. She had no known drug allergies. She was a married with two healthy daughters. She was working full time in a medical office. Her family history was not contributory.
Examination revealed normal vital signs. She had bilateral moderate parotid enlargement which were tender to palpation (right more than left side), and only the right parotid gland was slightly red. Intraorally, saliva was expressed from Stensen’s and Whartin duct orifices. She did not complain of dry mouth or eyes. Temporomandibular joint examination was within normal limits. No other significant finding was noticed in intra/extra oral examination. Laboratory testing did not show any abnormality in the CBC, chemistries, liver profile, or blood glucose. Serology indicated significantly elevated ANA titer of more than 1:1280 (normal: <1:40), both anti-Ro and anti-La titers were more than 8.0 (normal: <1 by commercial ELISA). The erythrocyte sedimentation rate was 28 (normal: 20 mm/h or less). A biopsy of minor salivary gland was performed in the left inner lower lip under local anesthesia. The result indicated periductal lymphocytic infiltration with focus score of 12 (Figures 2(C) and (D)).
The patient was then seen in the Oklahoma Medical Research Foundation Sjögren’s Syndrome Clinic. Eye examination with Schirmer test and Lissamine green indicated dry eyes. She met international criteria for Sjögren’s syndrome [21]. At this time patient has occasional pain of the parotid glands. She has been taking Plaquenil 200 mg twice/day for past few years with improvement in joint pain.
3. Discussion
There are only a few cases regarding Kikuchi-Fujimoto disease and SS is reported. Kikuchi-Fujimoto disease is a relatively new condition but is it known worldwide. The patients are usually young adults with a slight predilection for female (55/23 = F/M ratio), and mean age of 30 years with a range of 11 - 75 years and more Caucasian than others were reported (60/6 = W/B) [9]. The most
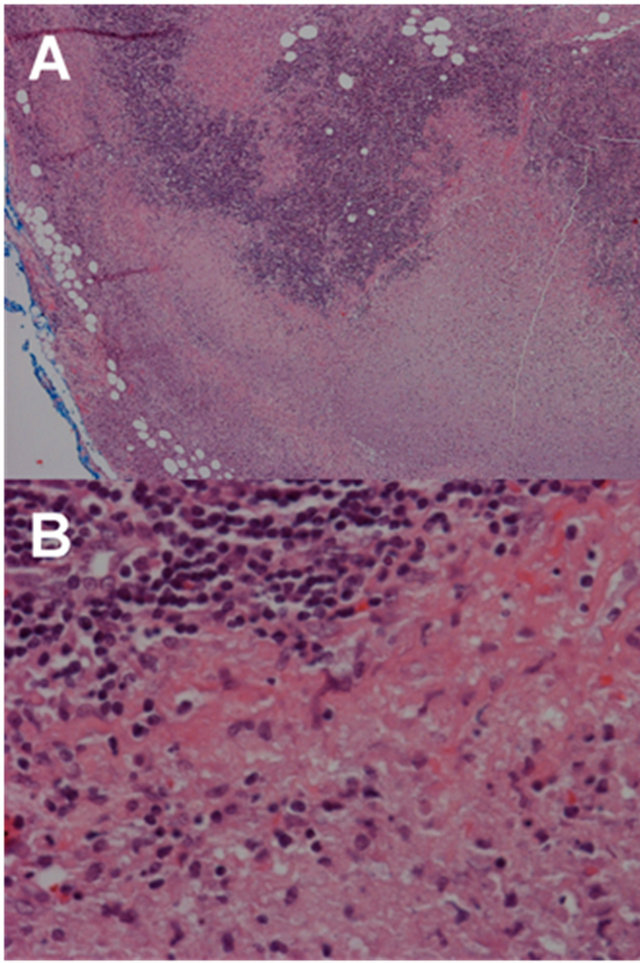
Figure 1. (A) Low-power image of the lymph node showing lymphoid tissue with area of necrosis (20× magnification); (B) High-power image of the same case showing necrosis with abundant histiocytes and lymphocytes and lack of neutrophills (400× magnification).
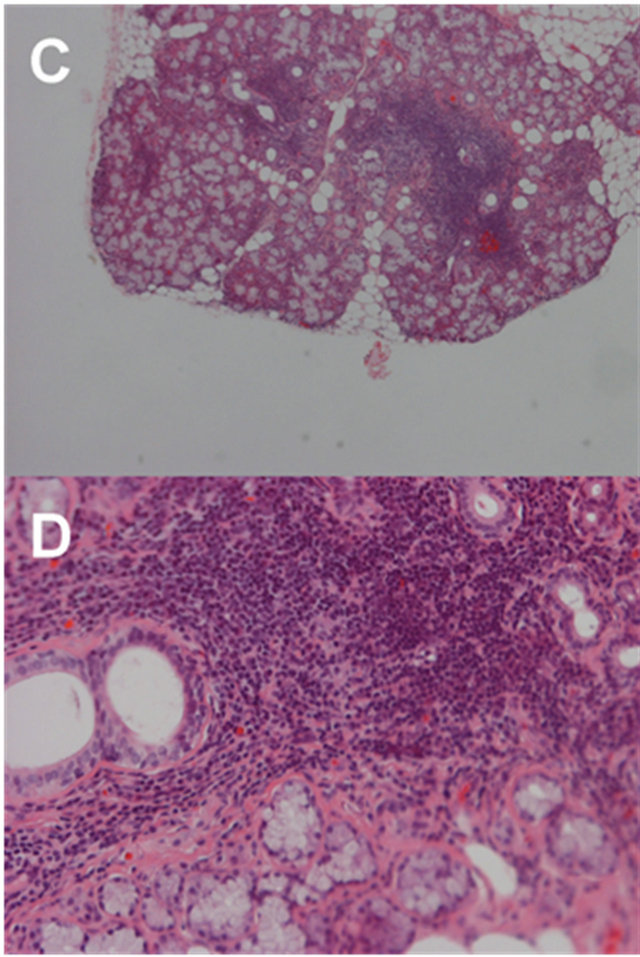
Figure 2. (C) Low-power image of the minor salivary gland showing foci of periductal lymphocytic infiltration (40× magnification); (D) High-power image of the same case showing periductal lymphocytic infiltration (200× magnification).
common sign is localized unilateral cervical lymphadenopathy. Common locations include axilla, supraclavicular, mediastinal, inguinal, intraparotid, iliac, celiac, and peripancreatic areas. The enlarged lymph nodes may be associated with pain, and some patients manifest systemic symptoms, including fever, myalgias, and skin rashes. The clinical course is typically benign, with resolution of the adenopathy in a few weeks to months [23].
Duration of symptoms are reported to be 1 - 24 months with a mean of 3 months [9]. Atypical peripheral blood lymphocytes were reported in some patients. Neutropenia, lymphocytosis, and thrombocytopenia were rarely reported [9,24].
Histological examination is valuable for an accurate diagnosis of Kikuchi-Fujimoto disease. The histopathologic characteristic of a lymph node in Kikuchi-Fujimoto disease include patchy necrosis of paracortical area, and abundant extracellular apoptotic nuclear debris. A mixture of different types of histiocytes include nonphagocytic histiocytes known as crescentic-shaped histiocytes as well as plasmacytoid monocytes and small lymphocytes surrounding the necrotic areas [24,25]. Histiocytic immunophenotypes are positive for CD68, lysozyme, and myeloperoxidase. T cells within the lesions are more likely to be cytotoxic (CD8+). Granulocytes such as neutrophils are absent and plasma cells are rare or absent [23,24].
Kikuchi-Fujimoto disease has been reported in association with SLE. Histopathologic features of KikuchiFujimoto disease and SLE may be similar. In both SLE and Kikuchi-Fujimoto disease, cellular protein structures have been found in the cytoplasm of lymphocytes and histiocytes that emphasize the hypothesis that Kikuchi-Fujimoto disease is a self-limited SLE-like disorder [26,27], but these two diseases can be differentiated by the absence of the neutrophils, plasma cells, and hematoxylin bodies in Kikuchi disease. Plasma cells are a prominent feature of SLE. Some authorities believe that Kikuchi-Fujimoto disease could be a precursor for SLE, as both diseases have had coexisting patterns in the same patients [12-20,23]. Some percentage of Kikuchi-Fujimoto disease patients go on to develop SLE, about 3% in one series [27], while a greater number (32 of 244, 13%) of reported patients with Kikuchi-Fujimoto also have SLE [20]. All variants of time course have been seen. That is, simultaneous onset, Kikuchi-Fujimoto disease preceding SLE, and SLE preceding Kikuchi-Fujimoto disease [19].
Sanpavat et al. [28] studied 40 cases of Thai patients who had been diagnosed as having necrotizing nongranulomatous lymphadenitis. The clinical features, histomorphology, and special stains were evaluated. Of those 40 patients, 17 (42.5%) had Kikuchi-Fujimoto disease, 8 (20%) had tuberculosis (TB) lymphadenitis, and 1 (2.5%) had lymphadenitis associated with SLE. Due to a lack of follow-up data, 14 (35%) did not have a specific diagnosis [28]. Of course, tuberculosis will be much less common in other areas of the world. Nonetheless, this report does give an estimate consistent with other reports of the frequency of SLE in patients with necrotizing lymphadenitis.
A report in Chinese reported two patients, a 14-yearold boy with SLE and secondary Sjögren’s syndrome with 8 months disease duration, and a 9-year-old boy with primary Sjögren’s syndrome who was diagnosed after 3 years of disease duration. Both had multi-organ involvement and biopsy proven KFD [29].
Our patient had Kikuchi-Fujimoto disease followed a few years later with Sjögren’s syndrome. Given the known association of SLE to Kikuchi-Fujimoto disease, it is perhaps not surprising that Sjögren’s syndrome may also be associated. SLE and Sjögren’s syndrome are clearly related, both clinically and serologically. While dry eyes and dry mouth are the hallmark manifestations of Sjögren’s syndrome, these patients, similar to those with SLE may have non-deforming inflammatory arthritis, vasculitic skin rash, photosensitivity as well as lymphopenia and leukopenia [30,31]. Anti-Ro and anti-La are found in both diseases, albeit more commonly in those with Sjögren’s syndrome [32]. Three other patients have been reported with Kikuchi-Fujimoto and Sjögren’s syndrome (Table 1). As described with SLE, KikuchiFujimoto disease has preceded, been diagnosed simultaneously with and occurred after Sjögren’s syndrome. All the reported patients have had anti-Ro and anti-La, consisted with a large amount of data that manifestations of Sjögren’s syndrome beyond the lacrimal and salivary glands are associated with the presence of these autoantibodies.
The differential diagnosis of patients presenting with KFD includes lymphoma. The presence of the large plasmacytoid monocytes in KFD may be mistaken for lymphoma cells. But, a mixture of lymphocytes and histiocytes and the presence of reactive follicles can help to separate these two diseases [9,24].
Table 1. Reported patients with Kikuchi-Fujimoto disease and Sjögren’s syndrome in English. The report of Lu, et al. is in Chinese.

In summary, KFD is a rare, but perhaps under-recognized disease. It can easily be confused with lymphoma and lymphadenopathy in SLE. To date, only a few cases of KFD with Sjögren’s syndrome have been reported in English literature [33,34]. Although KFD is a self-limiting condition, patients should be followed for the possibility of developing autoimmune disease, including SS and SLE.
REFERENCES
- N. Talal and J. J. Bunim, “The Development of Malignant Lymphoma in the Course of Sjögren’s Syndrome,” The American Journal of Medicine, Vol. 36, No. 4, 1964, pp. 529-540. doi:10.1016/0002-9343(64)90101-9
- A. G. Tzioufas, “B-Cell Lymphoproliferation in Primary Sjogren’s Syndrome,” Clinical and Experimental Rheumatology, Vol. 14, Suppl. 14, 1996, pp. S65-S70.
- J. M. Anaya, G. J. Tobon, P. Vega and J. Castiblanco, “Autoimmune Disease Aggregation in Families with Primary Sjögren’s Syndrome,” The Journal of Rheumatology, Vol. 33, No. 11, 2006, pp. 2227-2234.
- J. E. Moyron-Quiroz, J. Rangel-Moreno, K. Kusser, L. Hartson, F. Spraque, S. Goodrich, D. L. Woodland, F. E. Lund and T. D. Randall, “Role of Inducible Bronchus Associated Lymphoid Tissue (iBALT) in Respiratory Immunity,” Nature Medicine, Vol. 10, No. 9, 2004, pp. 927- 934. doi:10.1038/nm1091
- S. S. Kassan, T. L. Thomas, H. M. Moutsopoulos, R. Hoover, R. P. Kimberly, D. R. Budman, J. Costa, J. L. Decker and T. M. Chused, “Increased Risk of Lymphoma in Sicca Syndrome,” Annals of Internal Medicine, Vol. 89, No. 6, 1978, pp. 888-892.
- M. Voulgarelis, U. G. Dafni, D. A. Isenberg and H. M. Moutsopoulos, “Malignant Lymphoma in Primary Sjögren’s Syndrome: A Multicenter, Retrospective, Clinical Study by the European Concerted Action on Sjögren’s Syndrome,” Arthritis and Rheumatism, Vol. 42, No. 8, 1999, pp. 1765-1772. doi:10.1002/1529-0131(199908)42:8<1765::AID-ANR28>3.0.CO;2-V
- M. Kikuchi, “Lymphadenitis Showing Focal Reticulum Cell Hyperplasia with Nuclear Debris and Phagocytosis,” Nippon Ketsueki Gakkai Zasshi, Vol. 35, 1972, pp. 379- 380, Japanese.
- Y. Fujimoto, Y. Kozima and K. Yamaguchi, “Cervical Subacute Necrotising Lymphadenitis: A New Clinicopathologic Entity,” Naika, Vol. 20, 1972, pp. 920-927, (Japanese).
- R. F. Dorfman, G. J. Berry, “Kikuchi’s Histiocytic Necrotizing Lymphadenitis: An Analysis of 108 Cases with Emphasis on Differential Diagnosis,” Seminars in Diagnostic Pathology, Vol. 5, No. 4, 1988, pp. 329-345.
- S. D. Hudnall, T. Chen, S. Amr, K. H. Young and K. Henry, “Detection of Human Herpesvirus DNA in Kikuchi-Fujimoto Disease and Reactive Lymphoid Hyperplasia,” International Journal of Clinical and Experimental Pathology, Vol. 1, No. 4, 2008, pp. 362-368.
- H. C. Hollingsworth, S. C. Peiper, L. M. Weiss, M. Raffeld and E. S. Jaffe, “An Investigation of the Viral Pathogenesis of Kikuchi-Fujimoto Disease. Lack of Evidence for Epstein-Barr Virus or Human Herpesvirus Type 6 as the Causative Agents,” Archives of Pathology and Laboratory Medicine, Vol. 118, No. 2, 1994, pp. 134-140.
- G. Hedia, A. Jamel, A. Maher, A. Hanadi, H. Agnes and K. Nidhameddine, “Kikuchi-Fujimoto Disease Associated with Systemic Lupus Erythematosus,” Journal of Clinical Rheumatology, Vol. 11, No. 6, 2005, pp. 341-342. doi:10.1097/01.rhu.0000191539.52897.ee
- O. Meyer, M. F. Kahn, M. Grossin, P. Ribard, N. Belmatoug, F. Morinet and J. C. Fournet, “Parvovirus B19 Infection Can Induce Histiocytic Necrotizing Lymphadenitis (Kikuchi’s Disease) Associated with Systemic Lupus Erythematosus,” Lupus, Vol. 1, No. 1, 1991, pp. 37-41. doi:10.1177/096120339100100107
- B. Tumiati, A. Bellelli, I. Portioli and S. Prandi, “Kikuchi’s Disease in Systemic Lupus Erythematosus: An Independent or Dependent Event?” Clinical Rheumatology, Vol. 10, No. 1, 1991, pp. 90-93. doi:10.1007/BF02208041
- M. D. Eisner, J. Amory, B. Mullaney, L. Tierney and W. S. Browner, “Necrotizing Lymphadenitis Associated with Systemic Lupus Erythematosus,” Seminars in Arthritis and Rheumatism, Vol. 26, No. 1, 1996, pp. 477-482. doi:10.1016/S0049-0172(96)80028-X
- S. Kaur, G. P. Thami and A. J. Kanwar, “Kikuchi’s Disease, Skin and Systemic Lupus Erythematosus,” The British Journal of Dermatology, Vol. 146, No. 1, 2002, pp. 167-168. doi:10.1046/j.1365-2133.2002.4513_4.x
- C. Martinez-Vazquez, G. Hughes, J. Bordon, J. AlonsoAlonso, A. Anibarro-Garcia, E. Redondo-Martinez and F. Touza-Rey, “Histiocytic Necrotizing Lymphadenitis, Kikuchi-Fujimoto’s Disease, Associated with Systemic Lupus Erythematosus,” Oxford Journals Medicine, Vol. 90, No. 8, 1997, pp. 531-533. doi:10.1093/qjmed/90.8.531
- G. A. Chamulak, R. K. Brynes and B. N. Nathwani, “Kikuchi-Fujimoto Disease Mimicking Malignant Lymphoma,” American Journal of Surgical Pathology, Vol. 14, No. 6, 1990, pp. 514-523. doi:10.1097/00000478-199006000-00002
- H. C. Lin, C. Y. Su, C. C. Huang, C. F. Hwang and C. Y. Chien, “Kikuchi’s Disease: A Review and Analysis of 61 Cases,” Otolaryngology Head and Neck Surgery, Vol. 128, No. 5, 2003, pp. 650-653. doi:10.1016/S0194-5998(02)23291-X
- Y. Kucukardali, E. Solmazgul, E. Kunter, O. Oncul, S. Yildirim and M. Kaplan, “Kikuchi-Fujimoto Disease: Analysis of 244 Cases,” Clinical Rheumatology, Vol. 26, No. 1, 2007, pp. 50-54. doi:10.1007/s10067-006-0230-5
- E. M. Tan, A. S. Cohen, J. F. Fries, A. T. Masi, D. J. McShane, N. F. Rothfield, J. G. Schaller, N. Talal and R. J. Winchester, “The 1982 Revised Criteria for the Classification of Systemic Lupus Erythematosus,” Arthritis and Rheumatism, Vol. 25, No. 11, 1982, pp. 1271-1277. doi:10.1002/art.1780251101
- C. Vitali, S. Bombardieri, R. Jonsson, H. M. Moutsopoulos, E. L. Alexander, S. E. Carsons, T. E. Daniels, P. C. Fox, R. I. Fox, S. S. Kassan, S. R. Pillemer, N. Talal, M. H. Weisman, European Study Group on Classification Criteria for Sjogren’s Syndrome, “Classification Criteria for Sjögren’s Syndrome: A Revisal of the European Criteria Proposed by the American-European Consensus Group,” Annals of Rheumatic Diseases, Vol. 61, No. 6, 2002, pp. 554-558. doi:10.1136/ard.61.6.554
- D. J. Good and R. D. Gascoyne, “Atypical Lymphoid Hyperplasia Mimicking Lymphoma,” Hematology/Oncology Clinics of North America, Vol. 23, No. 4, 2009, pp. 729-745. doi:10.1016/j.hoc.2009.04.005
- R. F. Dorfman, “Histiocytic Necrotizing Lymphadenitis of Kikuchi and Fujimoto,” Archives of Pathology and Laboratory Medicine, Vol. 111, No. 11, 1987, pp. 1026-1029.
- X. Bosch, A. Guilabert, R. Miquel and E. Campo, “Enigmatic Kikuchi-Fujimoto Disease: A Comprehensive Review,” American Journal of Clinical Pathology, Vol. 122, No. 1, 2004, pp. 141-152. doi:10.1309/YF081L4TKYWVYVPQ
- M. Imamura, H. Ueno, A. Matsuura, H. Kamiya, T. Suzuki, K. Kikuchi and T. Onoe, “An Ultrastructural Study of Subacute Necrotizing Lymphadenitis,” The American Journal of Pathology, Vol. 107, No. 3, 1982, pp. 292-299.
- S. Paradela, J. Lorenzo, W. Martínez-Gómez, T. YebraPimentel, L. Valbuena and E. Fonseca, “Interface Dermatitis in Skin Lesions of Kikuchi-Fujimoto’s Disease: A Histopathological Marker of Evolution into Systemic Lupus Erythematosus?” Lupus, Vol. 17, No. 12, 2008, pp. 1127-1135. doi:10.1177/0961203308092161
- A. Sanpavat, P. Wannakrairot and T. Assanasen, “Necrotizing Non-Granulomatous Lymphadenitis: A Clinicopathologic Study of 40 Thai Patients,” The Southeast Asian Journal of Tropical Medicine and Public Health, Vol. 37, No. 3, 2006, pp. 563-570.
- S. Lu, J. Zhang, W. Zhou and X. M. Wang, “Histiocytic Necrotic Lymphadenitis as an Initial Presentation in Two Children with Sjogren’s Syndrome and/or Systemic Lupus Erthematous,” Chinese Journal of Contemporary Pediatrics, Vol. 12, No. 4, 2010, pp. 311-312, (Chinese).
- E. L. Alexander, F. C. Arnett, T. T. Provost and M. B. Stevens, “Sjogren’s Syndrome: Association of Anti-Ro (SS-A) Antibodies with Vasculitis, Hematologic Abnormalities, and Serologic Hyperreactivity,” Annals of Internal Medicine, Vol. 98, No. 2, 1983, pp. 155-159.
- M. Ramos-Casals, R. Solans, J. Rosas, M. T. Camps, A. Gil, J. Del Pino-Montes, J. Calvo-Alen, J. JimenezAlonso, M. L. Mico, J. Beltran, R. Belenguer, L. Pallares, GEMESS Study Group, “Primary Sjogren Syndrome in Spain: Clinical and Immunologic Expression in 1010 Patients,” Medicine (Baltimore), Vol. 87, No. 4, 2008, pp. 210-219. doi:10.1097/MD.0b013e318181e6af
- M. Reichlin and R. H. Scofield, “Ro (SS-A) Antibodies,” In: Y Shoenfeld, ME Gershwin and PL Meroni, Eds., Textbook of Autoantibodies, 2nd Edition, Elsevier, Amsterdam, 2006, pp. 783-788.
- M. Soy, H. Peynirci, S. Bilgi, M. K. Adali and S. Guresci, “Kikuchi-Fujimoto Coexisted with Sjogren’s Syndrome,” Clinical Rheumatology, Vol. 26, No. 4, 2007, pp. 607-608. doi:10.1007/s10067-005-0184-z
- Y. Miyashita, M. Yamaguchi and W. Fujimoto, “Painful Indurated Erythema Suggestive of Kikuchi-Fujimoto Disease in a Patient with Primary Sjögren’s Syndrome,” The Journal of Dermatology, Vol. 30, No. 8, 2003, pp. 608- 611.