Open Journal of Stomatology
Vol.4 No.1(2014), Article ID:42278,4 pages DOI:10.4236/ojst.2014.41006
Nevoid basal cell carcinoma: Case report
1Department of Oral Surgery, Clinical Stomatological University Center of Kosova, Pristina, Kosovo
2Department of Maxillofaxial Surgery, Clinical University Center of Kosova, Pristina, Kosovo
3Department of Pedodontics and Preventive Dentistry, Clinical Stomatological University Center of Kosova, Pristina, Kosovo
Email: *zana.agani@gmail.com
Copyright © 2014 Zana Agani et al. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited. In accordance of the Creative Commons Attribution License all Copyrights © 2014 are reserved for SCIRP and the owner of the intellectual property Zana Agani et al. All Copyright © 2014 are guarded by law and by SCIRP as a guardian.
Received 3 December 2013; revised 4 January 2014; accepted 16 January 2014
ABSTRACT
Nevoid basal cell carcinoma, known as Gorlin Goltz Syndrome, is a rare hereditary condition, characterized by a wide range of developmental abnormalities and a predisposition to neoplasms. In this report, we discuss a case of a patient with Gorlin Goltz Syndrome, who was 16 years old when first admitted for an initial appointment. The patient was diagnosed, treated and followed up for 7 years to present day. This syndrome is associated with a broad spectrum of anomalies and neoplasms as basal cell carcinomas, odontogenic keratocysts, palmar and/or plantar pits, and ectopic calcifications of the falx cerebri. It affects multiple organ systems, which include skeletal, teeth, jaw, skin, eyes, reproductive organs, and neural system. All the features however, are rarely observed in a single patient. The following paper presents the significance of early diagnosis of Gorlin Goltz Syndrome and the importance of a multidisciplinary approach in providing proper treatment for the patient.
KEYWORDS
Nevoid Basal Cell; Gorlin Goltz; Keratocyst
1. INTRODUCTION
Gorlin-Goltz syndrome is a hereditary condition (disorder) which is transmitted as an autosomal dominant trait and characterized with a defect or mutation in 9th chromosome [1]. Jarich and White made the first description of patients with this syndrome in 1894, by highlighting the presence of multiple basocellular carcinomas. Decades later in 1960, Gorlin and Goltz established a classical triad with multiple basocellular carcinomas, keratocyst of the jaw and bifid ribs [2]. There are different synonyms for this condition known as nevoid basal cell carcinoma syndrome, Gorlin Syndrome, Gorlin Goltz syndrome, and basal cell nevus syndrome fifth pacomatosis [3]. The estimated prevalence varies from 1/57,000 to 1/256,000 among large number of studies, with a male-to-female ratio of 1:1 [4]. The differential diagnosis should be done with Bazex syndrome, Trichoepitelioma papulosum multiplex and Torre’s syndrome [3].
The early diagnosis of this syndrome combined with a multi-specialist team consulting (oral and maxillofacial surgeon, dermatologist, neurologist and genetics) is mandatory in order to establish proper treatment, while regular follow-ups are important to lower the risks of possible complications which may follow the syndrome.
The objective of this study is to emphasise the importance of early diagnosis in order to provide a better prognosis to the patient.
2. CASE REPORT
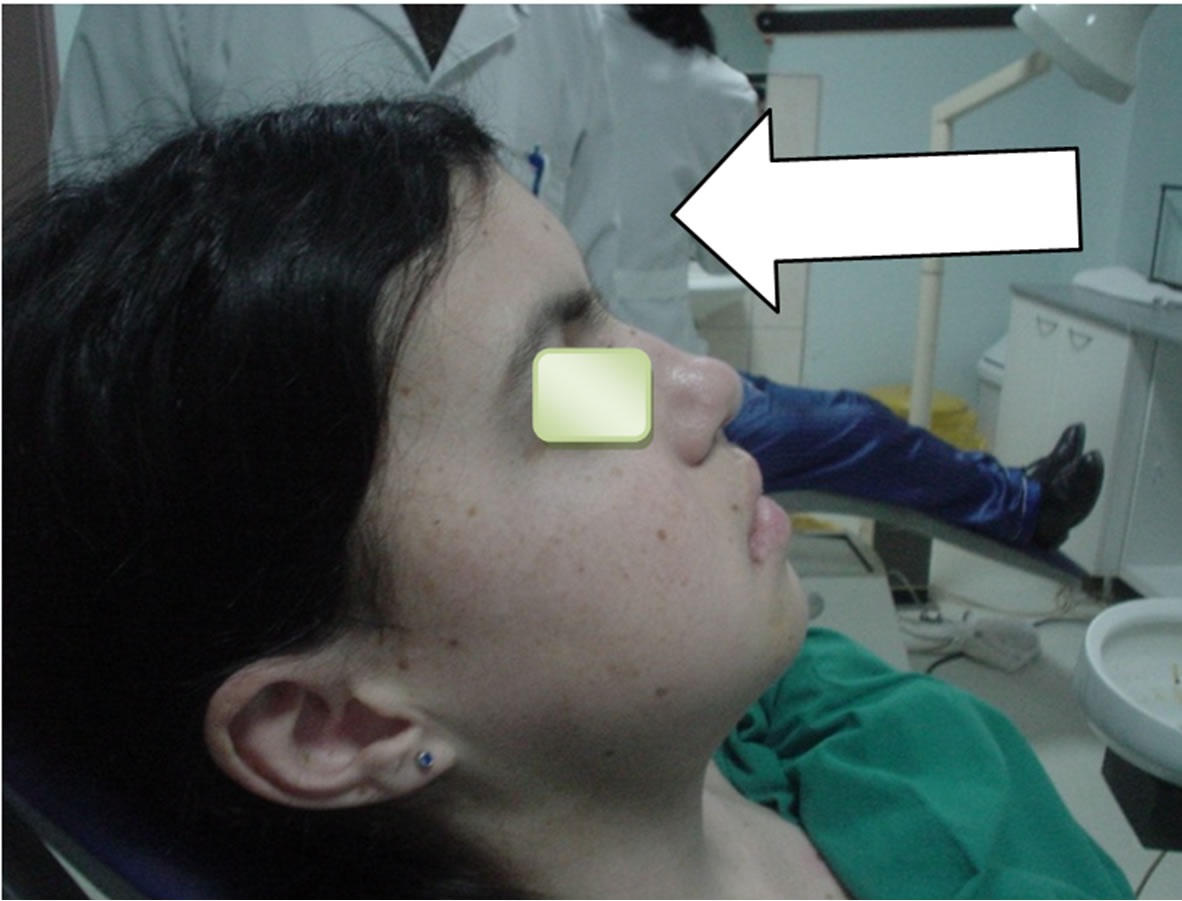
A 16-year-old female patient was admitted due to the unpainful deformity (swelling) presented in the frontal maxillary region. We took anamnesis, conducted a clinical examination and orthopantomography. Orthopanotomography revealed multiple well-defined radiolucencies with sclerotic border located in maxilla and mandible (Figure 1). The clinical extraoral examination (Figure 2) and the multiple well defined radiolucencies—cysts raised suspicions about the possibility of GGS, therefore continued relevant examinations were conducted.
By extraoral examination we saw eyes which appear wider apart than usual and well defined frontal bossing (Figure 2). The Craniofacial RTG did not confirm the calcification of falx cerebri (Figure 3) but the RTG of
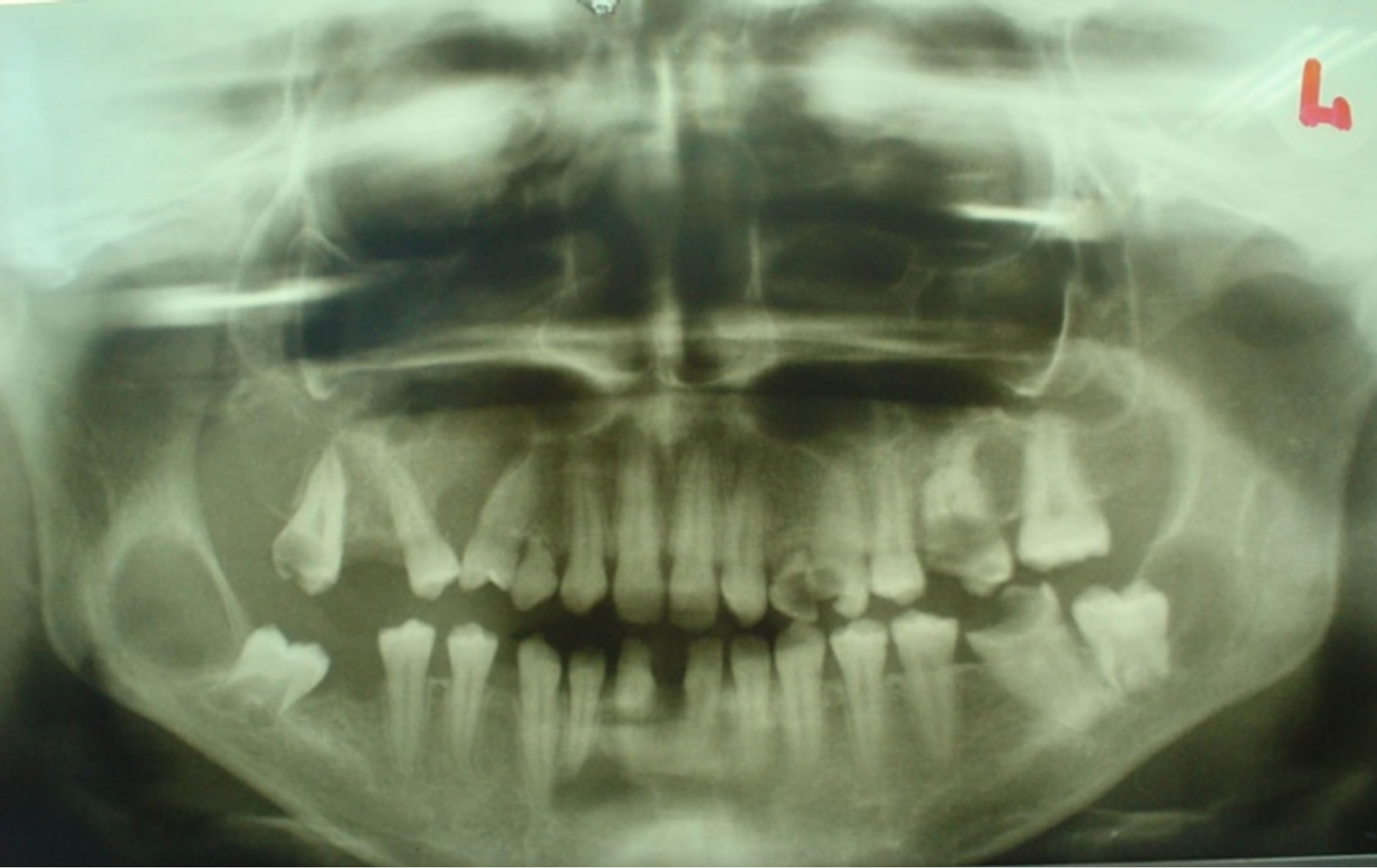
Figure 1. Orthopantomograph showing multiple multilocular well defined radiolucencies with sclerotic border present in both maxilla and mandible.
 (a)
(a)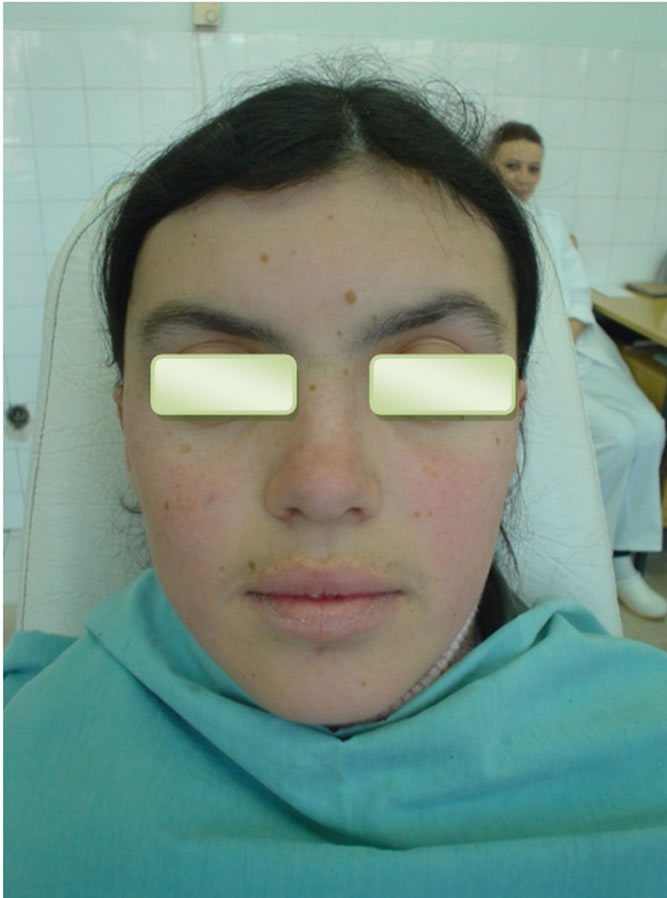 (b)
(b)
Figure 2. Figure’s shows eyes wide apart, frontal bossing and broad nasal bridge.
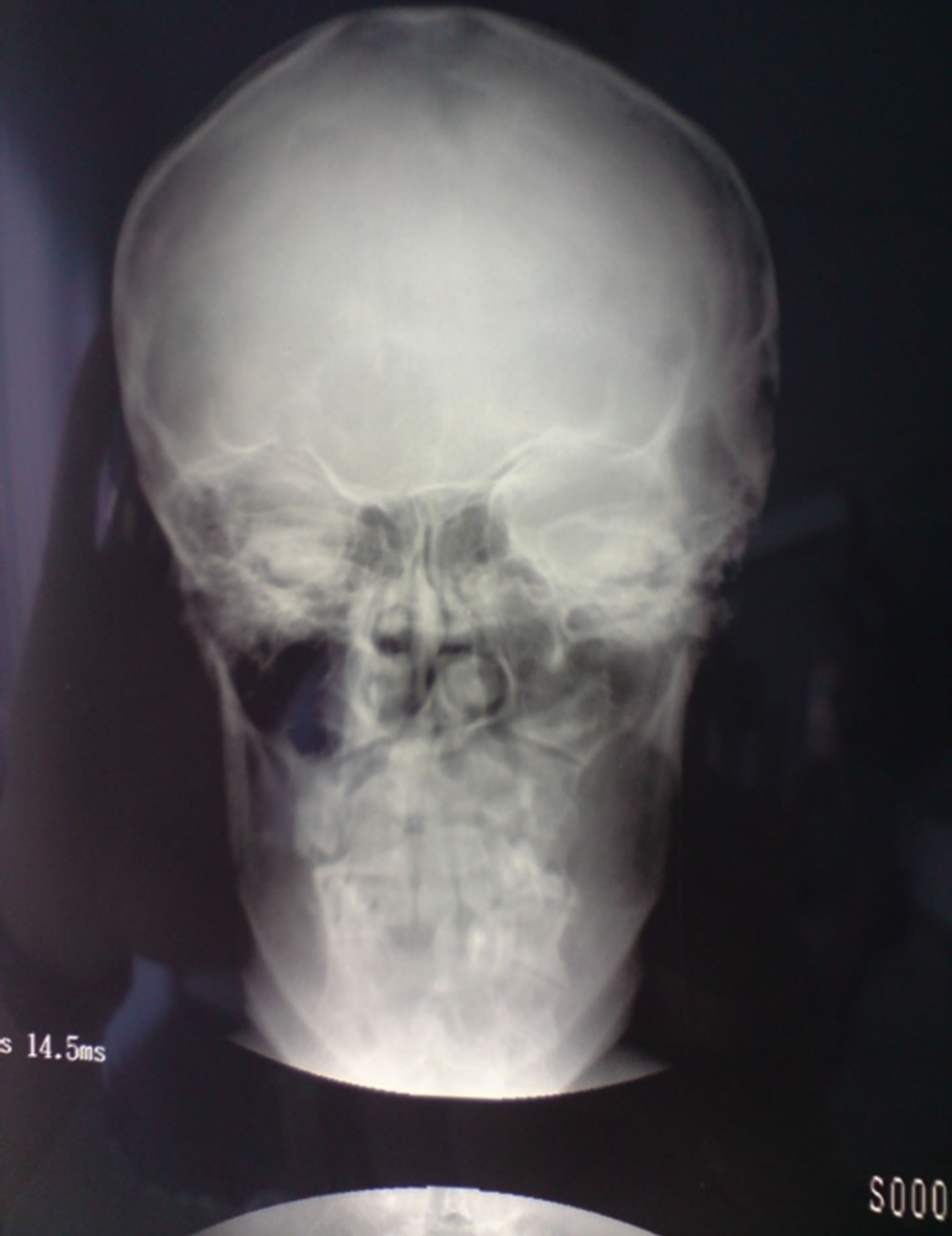
Figure 3. Craniofacial RTG without signs of calcification.
the spinal cord confirmed the presence of skeletal deformity-scoliosis (Figure 4).
Based on the findings, an enuclation of all cysts pre-
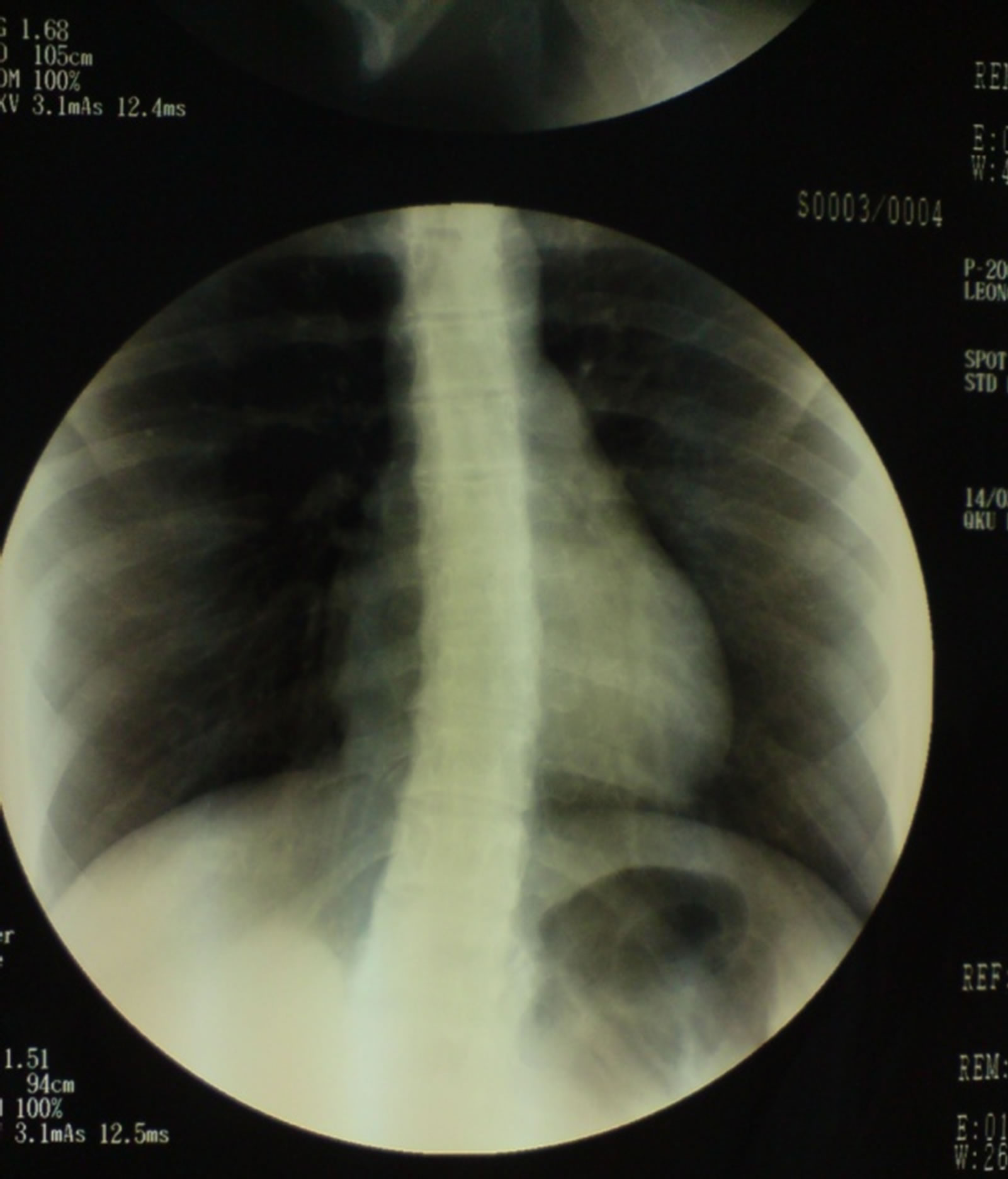
Figure 4. RTG of chest showing a signs of thoracic scoliosis.
sented was conducted, with the extraction of the tooth without bone support. The histopatology finding confirmed the hiperceratinised tissue with characteristics of keratocyst (Figure 5).
The patient was well informed about the characteristics of the syndrome. In this case, regular follow-ups were maintained in order to control the possible changes that may occur in the jaw, with treatments conducted in order to minimize eventual complications. The patient was advised to avoid sun exposure and use protective creams with high sun block factors, in order to protect the skin from possible neoplastic changes.
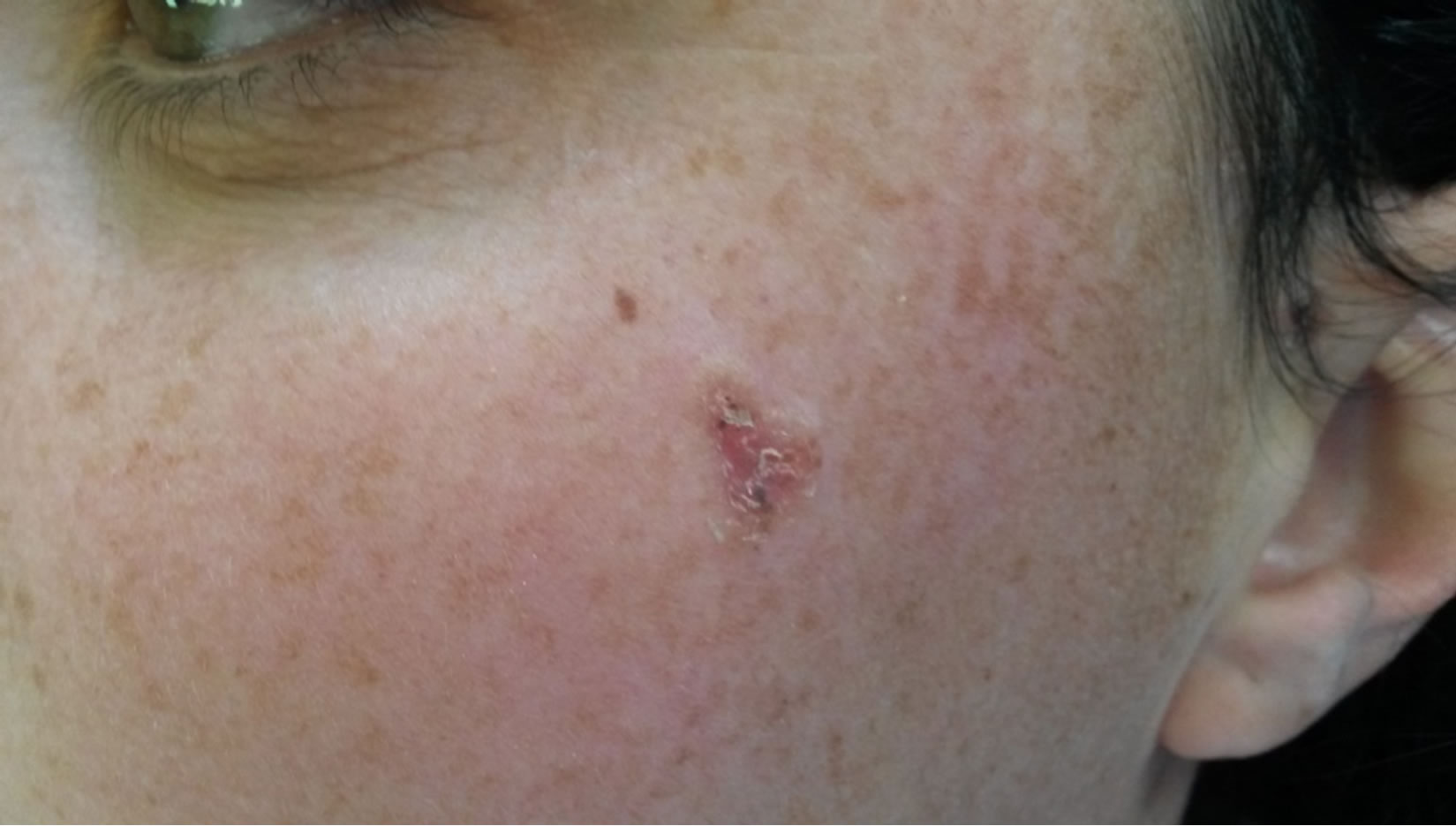
After the first initial appointment seven years prior, the first notable changes in skin susceptible to basal cell carcinomas (Figures 6(a)-(c)), after surgical treatment and a histopatological exam were confirmed (Figure 7). Furthermore, a DNA analyses was advised, in order to confirm the diagnosis on the gene level.
3. DISCUSSION
Nevoid basal cell carcinoma syndrome (NBCCS), or also called Gorlin Goltz Syndrome is a hereditary condition transmitted as an autosomal dominant trait with complete penetrance and variable expressivity. The syndrome is characterised by numerous basal cell carcinomas (BCCs), odontogenic keratocysts of the jaw, palmar and/or plantar pits, skeletal abnormalities and intracranial calcifications [5].
Basal cell carcinomas may arise in various stages of the syndrome: most often they appear between puberty and 35 years of age [3]. The Nevoid basal cell carcinomas are unlikely to occur under the age of puberty unless the patient has been exposed to radiotherapy, but cases as early
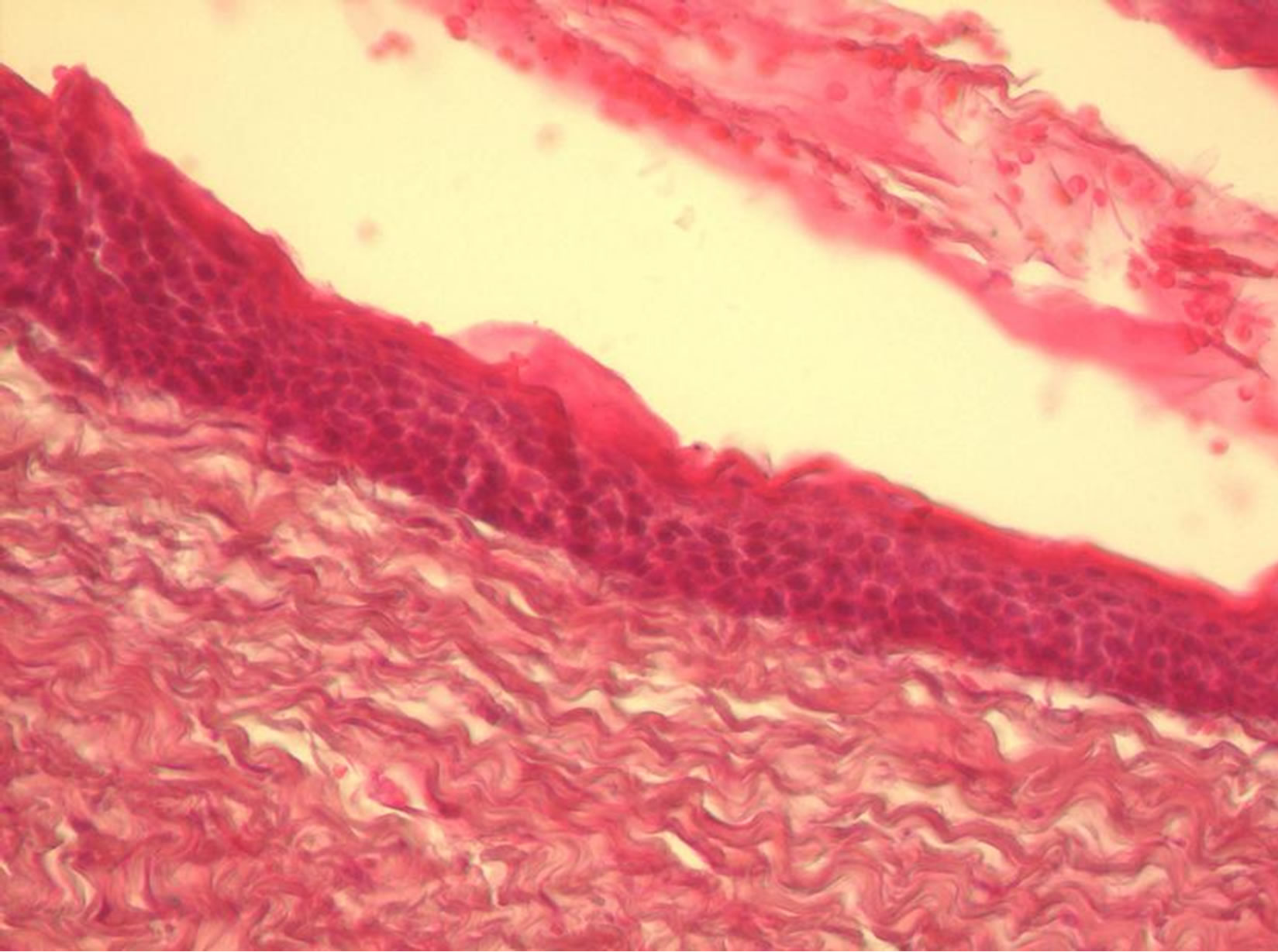
Figure 5. The histopathology finding confirms for keratocysts.
 (a)
(a)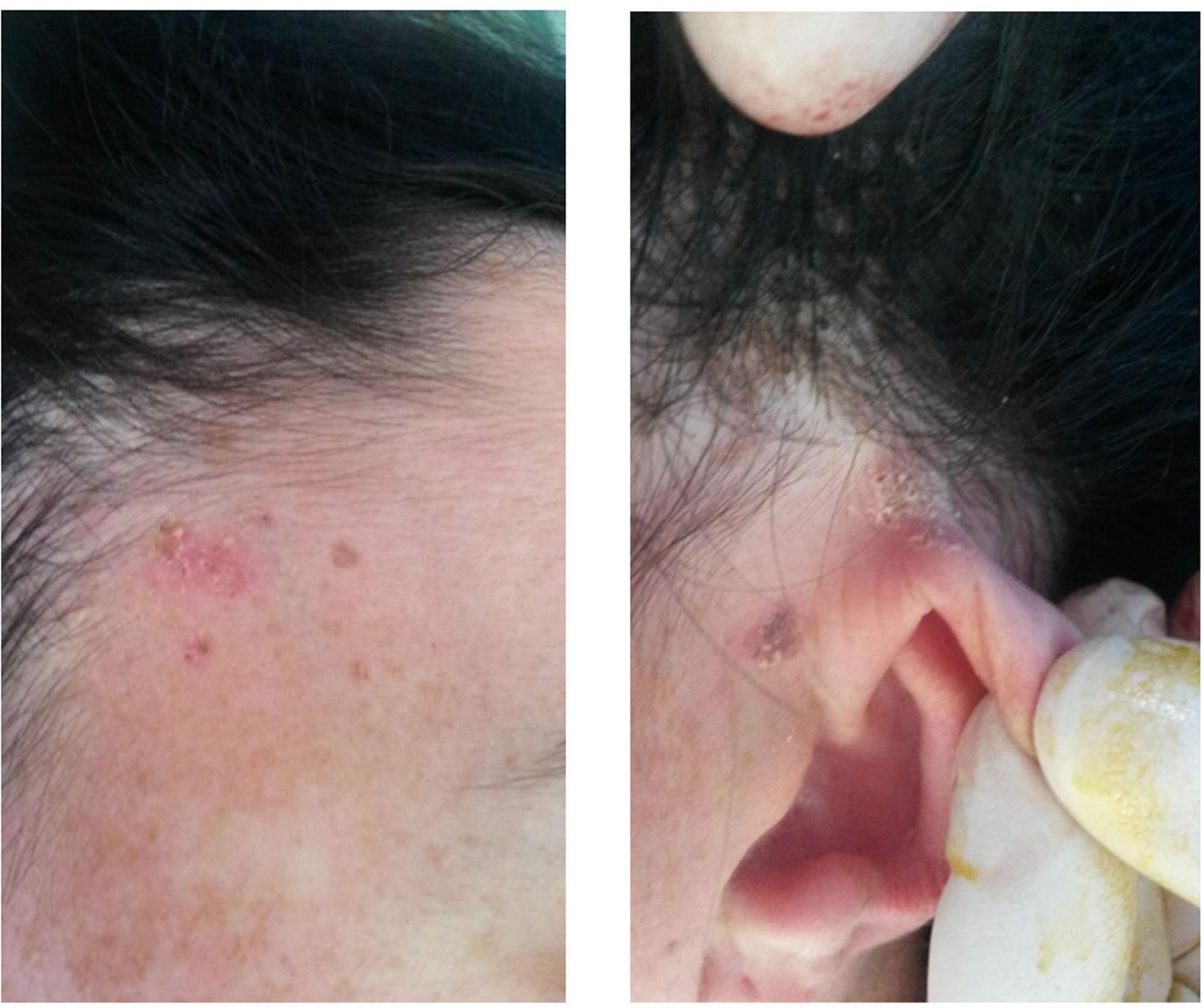 (b)
(b)
Figure 6. (a) BCC-confirmed with histopathology in zygomatic region; (b) BCC-temporal region; (c) BCC in preauricular region.
as 2 years have been described [6].
Odontogenic keratocyst (OKC) also known as keratinizing odontogenic tumor (KOT), is the cystic lesion of odontogenic origin that demonstrates the behaviour of a benign neoplasm with propensity recurring following surgical treatment. Based on the clinical behaviour and molecular biology of the KOT, the terminology has prompted a change from OKC and led to its recent classification as an odontogenic tumor (OT). In the most
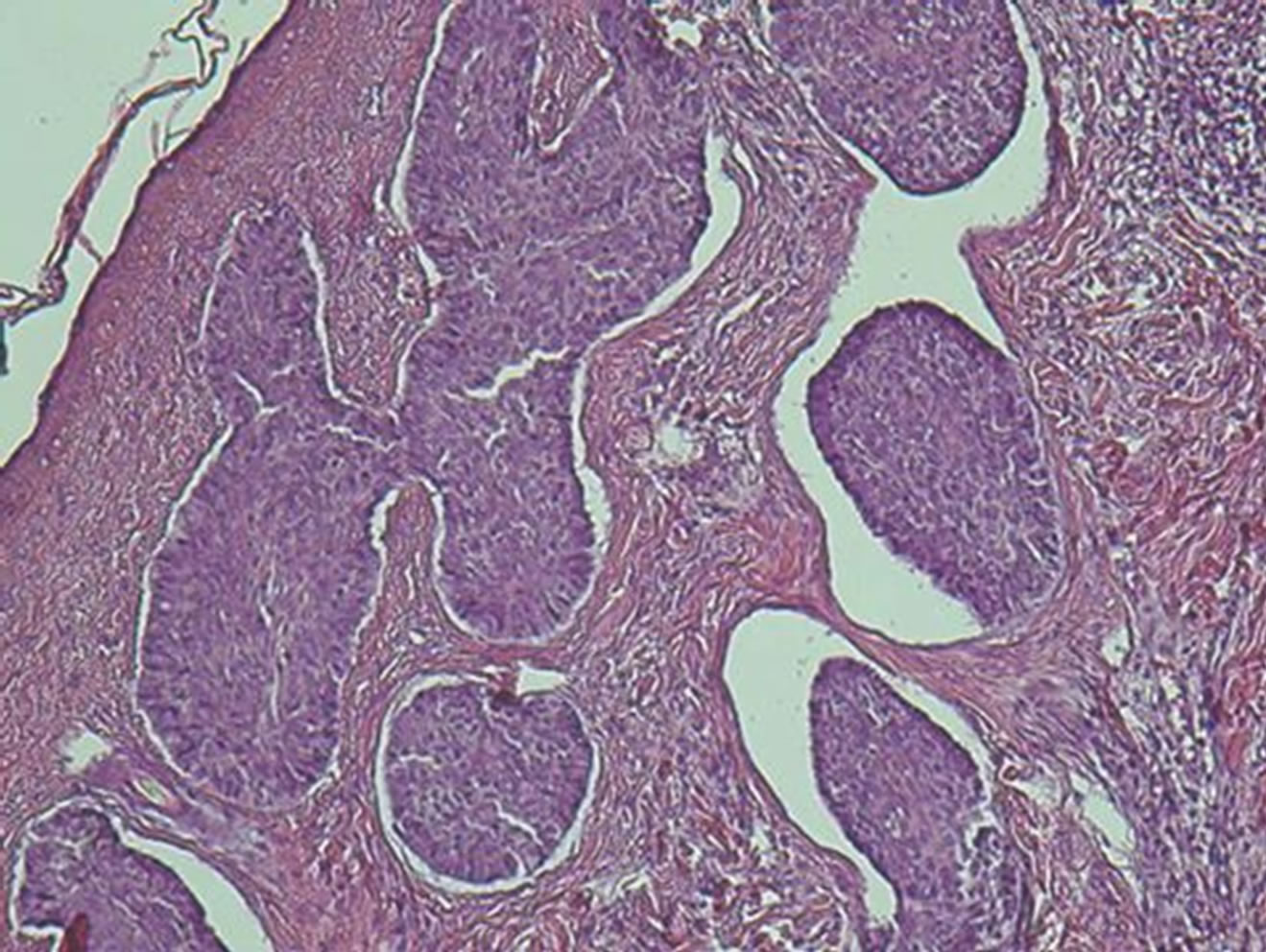
Figure 7. The histopathology finding confirms for Basal cell carcinomas.
recent WHO classification, the OKC has been identified as OT, therefore named keratocystic odontogenic tumor [7]. Keratocysts may occur as a part of NBCCS and commonly recur after treatment with recurrence rate of up to 60% [8]. They are rarely symptomatic and can rise by losing the bone around the tooth which leads to the loss of bone and pathological fractures [6]. Less than 10% of patients with multiple OKC have other manifestations of this syndrome. It has been suggested however, that multiple OKC alone may be the confirmatory of the syndrome [2]. Due to the abnormal growth of the skull, about 70% of patients affected by NBCCS have eyes which appear wider apart than usual. Other skeletal signs are scoliosis (40%), and abnormalities such as bifid, wide, fused, partially missing or underdeveloped ribs (30% - 60%) [5]. Evans et al. established the diagnostic criteria for NBCCs while Kimonis et al. in 1973 modified it. According to them, diagnosis of Gorlin-Goltz Syndrome can be established when two major or one major and two minor criteria are present which are described below:
Major criteria
1) Multiple (>2) BCCs or one BCC under the age of 20 years.
2) Odontogenic keratocyst of the jaw histologically proven.
3) Cutaneous palmar or plantar pits (≥3).
4) Bifid, fused or markedly splayed ribs.
5) Billameral calcification of the falx cerebri.
6) First degree relative with nevoid basal cell carcinomas.
Minor criteria1) Macrocephaly proven after adjustment for height.
2) Orofacial congenital malformations (≥1): cleft lip or palate, frontal bossing, “coarse face”, moderate or severe hypertelorism.
3) Other skeletal abnormalities: sprengel deformity, marked pectus deformity, marked syndactyly of the digits.
4) Radiological abnormalities: Bridging of the sella turcica, vertebral anomalies such as hemivertebrae, fusion or elongation of the vertebral bodies, modeling defects of the hands and feet, or flame shaped lucencies of the hands or feet.
5) Ovarian fibroma.
6) Medulloblastoma [9].
In our case we did conclude the presence of the syndrome based on 2 major criteria (histologically proven OKC and more than two BCCs) and three minor criteria (proven moderate hypertelorism, frontal bossing and other skeletal abnormalities as scoliosis).
4. CONCLUSION
Gorlin-Goltz syndrome is an infrequent multi-systemic disease that is inherited in a dominant autosomal way and shows a variable expressiveness. In order to be able to arrive at an early diagnosis of the syndrome, at the very first suspicion, clinical and radiographic tests are recommended and treatment decided by a multidisciplinary team, composed of oral and maxillofacial surgeons, as well as dermatologist, neurologist and geneticist. And definitely, the regular follow-up is mandatory to prevent the risks of possible complications which may follow the syndrome.
CONFLICT OF INTEREST
Authors declare no conflict of interest.
REFERENCES
- Gorlin, R.J. and Goltz, R.W. (1960) Multiple novoid basal-cell epithelioma, jaw cysts and bifid rib. A syndrome. New England Journal of Medicine, 262, 908. http://dx.doi.org/10.1056/NEJM196005052621803
- Agrawal, A., Murari, A., Vutukuri, S. and Singh, A. (2012) Case report, Gorlin-Goltz syndrome: Case report of a rare hereditary disorder, case reports in dentistry.
- Lo Muzio, L. (2008) Nevoid basal ceel carcinoma syndrome (gorlin syndrome). Orphanet Journal of Rare Disease, 3, 32.
- Kalogeropoulou, Ch., Zampakis, P., Kazantzi, S., Kraniotis, P. and Mastronikolis, N.S. (2009) Gorlin-Goltz syndrome: Incidental finding on routine ct scan following car accident. Cases Journal, 2, 9087. http://dx.doi.org/10.1186/1757-1626-2-9087
- Lo Muzio, L., Nocini, P.F., Savoia, A., Consolo, U., Procaccini, M., Zelante, L., Pannone, G., Bucci, P., Dolci, M., Bambini, F., Solda, P. and Favia, G. (1999) Nevoid basal cell carcinoma syndrome Clinical findings in 37 Italian affected individuals. Clinical Genetics: An International Journal of Genetics Molecular and Personalised Medicine, 55, 34-40.
- Evans, D.G.R., Ladusans, E.J., Rimmer, S., Burnell, L.D., Thakker, N. and Farndon, P.A. (1993) Complications of the nevoid basal cell carcinoma syndrome: Results of a population based study. Journal of Medical Genetics, 30, 460-464.
- Anderson, L., Kahnber, K.E. and Pogrel, M. (2010) Oral and maxillofacial surgery. Wiley, Blackwell, Blackwell Publishing Ltd.
- Booth, P.W., Schender, A.S. and Hausmen, J.E. (1999) Maxillofacial Surgery, Volume 2. Churchil, Livingston, 1466- 1467
- Kohli, M., Kohli, M., Sharma, N., Siddiqui, S.R. and Tulsi, S.P.S. (2010) Gorlin-Goltz syndrome. National Journal of Maxillofacial Surgery, 1, 50-52.
NOTES
*Corresponding author.