Open Journal of Genetics
Vol.3 No.1(2013), Article ID:29011,12 pages DOI:10.4236/ojgen.2013.31006
Cell therapy of a patient with type III Osteogenesis imperfecta caused by mutation in COL1A2 gene and unstable collagen type I*
![]()
1Department of Transplantation, UJCM, Krakow, Poland
2Department of Pediatrics and Medical Genetics, Chair of Pediatrics, UJCM, Krakow, Poland
3Department of Clinical Immunology, UJCM, Krakow, Poland
4Department of General and Molecular Biology and Genetics, Medical University of Silesia, Katowice, Poland
5Network of CoE BioMedTech, Poland
Email: #mipietrz@cyf-kr.edu.pl, alsieron@sum.edu.pl
Received 25 January, 2013; revised 28 February, 2013; accepted 10 March, 2013
Keywords: Bone Mineralisation; Cell Therapy; Collagen Type I; Osteogenesis imperfecta; Triple Helix Stability
ABSTRACT
The allogenic bone marrow derived mesenchymal stem cells transplantation was given to the newborn girl diagnosed with osteogenesis imperfecta type III, with multiple bone fractures, extreme shortness and limbs deformities. The treatment was performed at the age of 4 and 6 weeks. The clinical diagnosis was supported by biochemical analysis of collagen type I recovered from culture medium of cultivated patient’s skin fibroblast, which revealed its triple helix instability at temperature about 2˚C lower than normal. Sequencing of both genes encoding procollagen type I revealed heterozygous substitution G23569A in COL1A2 gene causing change of glycine at position 517 to aspartate. The donor of mesenchymal stem cells was the girl’s father. She received two intravenous infusions of suspended cultured mesenchymal cells in 16 days apart without any side effects. An analysis of procollagen type I secreted to the culture medium by bone marrow-derived mesenchymal stem cells obtained from the patient, 3 months following transplantation revealed its normal triple helix stability. During the subsequent two years of follow up two new bone fractures were noted. Currently a twoyear-old girl’s presents extreme growth and weight deficiency. The motoric development is also retarded, but the patient constantly improves and makes progresses.
1. INTRODUCTION
Osteogenesis imperfecta (OI) is a genetic disease determined by autosomal dominant negative mutation of type I collagen, which is commonly know as “brittle bone disease”. The overall birth prevalence of OI is estimated to range between 1 in 20,000 and 1 in 30,000. Clinically OI is heterogenous disorder with wide range of manifestations, from very mild, through moderate, to fatally severe, including lethal cases [1]. It is a systemic disorder affecting primarily bones but also teeth and eyes as well as leading to progressive hearing impairment [2]. Bones in this condition are fragile such that affected individuals sustain fractures after very mild trauma and frequent fractures, from several to tenths are observed throughout the patients live [3]. Multiple skeletal features and poor healing is responsible for short stature, relative macrocephaly, scoliosis and bowing of the long bones. Two types of the OI are represented by most severe clinical outcome [4]. The type II OI, which is the most severe form of osteogenesis imperfecta and is almost uniformly lethal. This form of the disorder is characterised by extreme bone fragility, leading to fetal demise or death in perinatal period [5,6]. Second one in severity of clinical symptoms is type III of OI, which is leading to serious skeletal deformities and makes the patient to be wheelchaired or bed ridden. The frequency of type II and III osteogenesis imperfecta is estimated to be 1 in 10000.
The gold standard for postnatal diagnosis of OI is based on clinical and radiological features, confirmed subsequently by molecular diagnosis of collagen triple helix stability and detection of mutation [7]. In the majority of cases the cause of the OI is the mutation in one of the two genes encoding procollagen type I polypeptide chains, namely COL1A1 and COL1A2. In recent years, however, new evidence surfaced shading light on other genes, which when mutated might lead to autosomal recessive forms of OI although both procollagen genes do not show the mutation [8-11]. The incorrect genes both encoding procollagen type I and potential disturbances in its posttranslational processing are the major causes of poor quality of extracellular matrix, regardless lower collagen content or its poor stability.
Conventional management for patients with OI consisted of supplementation with vitamin D and minerals, or administration of bisphosphonates [8,12-15]. However, these methods did not ameliorate the symptoms of OI [12]. Moreover, in young teenage patients the bisphosphonates cannot be used continuously, because they slow down bone turnover and as consequence child’s growth and healing of eventual bone fractures [16]. Another option of supportive treatment of OI patients is surgical and orthopedic intervention or protection with the use of braces, steel rods and other orthopedic equipment [reviewed in 11]. Either, supportive treatment of OI did not provide sufficient clinical improvement, especially in the most severe forms. Neither the medications or the surgical treatment can lead to permanent amelioration of the OI symptoms. The obvious reason is the genetic nature of this disorder.
For patients with OI type II and III a cell or gene therapy could be offered as the only chance of life saving and prevention of severe disability. Already more than 10 years ago first clinical trials were conducted for treating the severe type of OI with cell therapy [17]. The trials were then further expanded on larger group of OI patients [18-20].
Here, we report a case of a girl with OI type III treated at the age of 4 and 6 weeks by transplantation of allogeneic bone marrow derived mesenchymal stem cells (MSC). The diagnosis of OI was established on the base of clinical and radiological features, and confirmed subsequently by molecular diagnosis which revealed procollagen thermal instability and detected a missense mutation in COL1A2 gene.
2. MATERIAL AND METHODS
2.1. Patient
A girl patient was born to a G2, P1 healthy mother at 39/40 weeks of gestation. A child was delivered by caesarean section, because of abnormal fetal heart rate pattern, pelvic position, and severe deformities of upper and lower extremities detected prior to the child’s birth during regular monitoring of pregnancy by ultrasound examinations. Both parents are healthy and unrelated individuals. The first pregnancy of this couple was a miscarriage. The analysis of family pedigree did not show any cases of OI or any other genetic diseases. The birth parameters of the patient were as follow: birth weight 2490 g, length 44 cm, head circumference 34 cm, Apgar score 3, 4, 5, 7 points. In delivery room the child was successfully resuscitated. The patient required support of ventilation (nasal CPAP) shortly after birth and during the first three days of life. Neither mechanical ventilation nor oxygen therapy were needed later on.
At the age of three days, the child was admitted to the Neonatal Intensive Care Unit of the Department of Pediatrics, Chair of Pediatrics, Jagiellonian University and hospitalized for subsequent three months. Clinical evaluation of the patient on admission revealed short stature, severe lower limb deformities, relative macrocephaly, triangular face and blue sclera. X-ray examination (baby-gram) showed 27 bone fractures, multiple deformities and reduced ossification of the skull calvarion. Based on the clinical and radiological findings of multiple bone fractures and severe limb deformities, the preliminary diagnosis of type II OI was made according to the classification by Sillence [21]. The anthropometric measurements were conducted and the dermatoglyphic patterns were analyzed. Genomic DNA was isolated from cultured fibroblasts for the molecular analysis of the COL1A1 and COL1A2 genes. The skin biopsy was performed to assess the stability of produced by fibroblasts collagen triple helix. The levels of serum bone turn-over markers (osteocalcin (OC), procollagen type I N-propeptide (P1NP), collagen type I crosslinked Ctelopeptide (Ctx)) were assessed. Then, on the basis of the natural cause of the disease, the result of molecular analysis revealing heterozygous mutation in exon 26 of COL1A2 gene and the diminished thermal stability procollagen triple helix, the diagnosis was changed to type III osteogenesis imperfecta. The patient was qualified to a clinical trial entitled “Cell therapy in treatment of the patients with type II and type III osteogenesis imperfecta”. The entry criteria for the study were as follow: 1) diagnosis of type II or III osteogenesis imperfecta, 2) parents agreement for participation in the clinical study, 3) approval of the Bioethics Committee of the Jagiellonian University (KBET/108/B/2007).
2.2. Patient’s Samples
The anthropometric measurements were conducted and the dermatoglyphic patterns were analyzed before mesenchymal cells transplantation was performed. A blood samples were taken to assess the level of serum bone markers (OC, P1NP, and Ctx). The skin biopsy was performed to assess the stability of collagen triple helix and genomic DNA purification for molecular analyses of COL1A1 and COL1A2 genes.
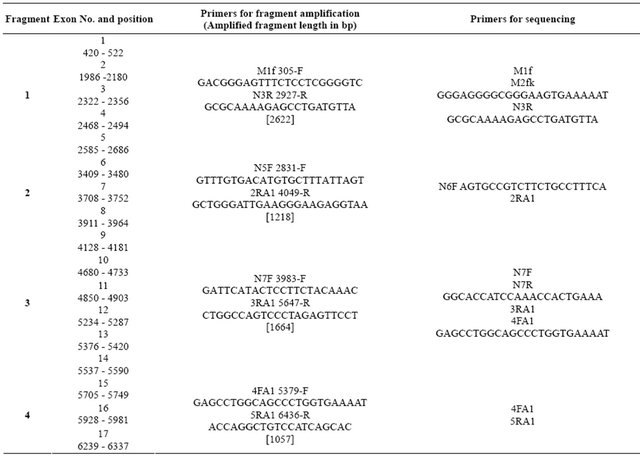
2.3. Survey of Collagen Genes for Mismatches
DNA fragments of the collagen COL1A1 and COL1A2 gene were amplified using primers described previously [22] and listed in Table 1. The primers were designed and verified using PrimerSelect in DNAStar programme package version 5.0. The DNA was isolated from the proband’s cultured fibroblasts, using BloodMini DNA purification kit (A & A Biotechnologies, Gdansk, Poland). The DNA was recovered from the kit column with 1 mM EDTA buffered with 10 mM Tris-HCl pH 8.5 at final volume 150 μl and its concentration was determined spectrophotometrically at the wave length 260 nm and 280 nm. Quality of the DNA was verified following its electrophoresis in 0.8% agarose gel. Until further analyses the DNA samples were stored at −20˚C. Concentra-tions of primers were calculated based on the sample absorbance at the wave length 260 nm and adjusted to the primer compositions. PCRs were performed according to the conditions described elsewhere [22]. Briefly, the reaction mixture included 300 ng double stranded patient genomic DNA, 1 µl 10 mM dNTPs (PCR nucleotide Mix, Roche, Germany), 1 µl (5 pmol) of each primer, 2.5 µl 10x Taq polymerase buffer, 1U Taq polymerase and nuclease free water (all from FastStart Taq DNA Polymerase, Roche, Germany) to the total volume of 25 µl. The mixture was denatured for 5 minutes at 95˚C, and incubated for 34 cycles of: 30 seconds at 95˚C, 30 seconds at 60˚C or 65˚C, depending of the Tm of the specific pair of primers (Table 1), 30 seconds to 2 minutes at 72˚C depending on the length of the amplified DNA fragment. When the last cycle was completed the samples were incubated for 7 minutes at 72˚C then cooled down and left at 4˚C. The PCR products (3 μl) were separated on 1% agarose gel (Agarose, LE, Analytical Grade, Promega, USA) in TAE buffer.
2.4. PCR Conditions for DNA Sequencing
The PCR for sequencing was conducted according to recommendations provided by the DNA Analyser 3130 × l ABI Prism, manufacturer (Applied Biosystems, USA). Briefly 10 to 20 ng of the PCR product was purified using Shrimp Alkaline Phospatase (Fermentas, USA) and Exonuclease I, E. coli (Fermentas, USA). Clean product was then combined in semi-skirted 96-well PCR plate with 5 pM of primer, 8 μl of the ready to use mixtureprovided by the manufacturer BigDye Terminator v3.1
Table 1. Sequences of primers used for fragment amplification and for sequencing of the COL1A1 human gene.
Continued

Cycle Sequencing Kit (Applied Biosystems, USA) containing polymerase, dideoxy-NTP in the buffer and the final volume was obtained by adjusting it with water up to 20 μl. The mixture was incubated in the thermocycler by Eppendorf for 35 cycles with the following steps in the single cycle: denaturation for 30 seconds at 96˚C, annealing 15 seconds at 50˚C, elongation for 3 minutes at 60˚C. After completing the last cycle the sample was cooled down to 4˚C. The sequencing PCR product were purified by BigDye XTerminator Purification Kit (Applied Biosystems, USA) according to manufacturers manual and put in the instrument for the DNA sequenceing. The sequences were compared with reference sequence (NG_007400.1 COL1A1 and NG_007405 COL1A2) using MegAlign in DNAStar programme package version 5.0. The differences were verified for final confirmation by analysis of the raw chromatogram sequencing data using Chromas Lite version 2.01 freeware.
2.5. Procollagen Type I Production and Purification
Procollagen type I was produced using skin fibroblasts prior and following the bone marrow mesenchymal stem cells transplantation. The skin biopsies of the size of 5 mm2 were obtained using surgical blade. The sample was minced into small pieces and placed on a 100 mm in diameter Petri dish containing 10 ml of Dulbeco’s Modified Eagle’s Medium supplemented with 4.5 g/L glucose, (high glucose DMEM) 10% foetal calf serum (FCS), 10,000 units penicillin, 10 mg units streptomycin, 25 μg/ml amphotericin B (all reagents from PAA The Cell Culture Company, Pasching, Austria). The medium was changed every other day. On day 7 of the culture the pieces of skin were removed and the fibroblasts were cultured until confluence. At 100% density the cells were transferred into T150 culture flask and maintained in the same medium until reaching 100% confluence when half of the culture was frozen, whereas, the other half was further cultured until 6 flasks of about 70% confluent were obtained. The procedure was followed for each sample. The fibroblast cultures were rinsed twice with sterile phosphate buffered saline (PBS) and filled with 15 ml of serum-free high glucose DMEM supplemented with ascorbic acid 50 μg/ml and all the other supplements as described above. The medium was collected every day and replaced with the fresh one for three consecutive days. Then for 24 hours the culture was fed on high glucose DMEM with serum. The procedure of washing with PBS, harvesting the serum-free medium and feeding with total complete medium were repeated 4 times.
Harvested medium was processed as previously described [23] with subsequent modifications described elsewhere [22,24]. The volume of sample containing procollagen I was reduced to the appropriate collagen concentration using Amicon cell under nitrogen pressure fitted with the membrane with the size cut off 100,000 kDa, following double dialysis against 200 volumes of so called storage buffer containing 0.4 M NaCl, 0.1 M Trizma Base (pH 7.4) supplemented with 0.025 M EDTA and 0.04% NaN3.
2.6. Preparation of Collagen Type I and Its Triple Helix Thermal Stability Assay
The procollagen type I sample was treated with acetic acid to adjust pH to about 2.0. Pepsin was added to the acidified procollagen to a final concentration 200 μg/ml. The digestion with pepsin was carried at 10˚C for 18 hours, than terminated by raising pH to 7.5. The concentration of collagen was determined using kit by Biocolor (Ireland). Stability of collagen triple helix was assessed using J-815 Spectropolarimeter (Jasco) at 220 nm, using a 0.5 mm path-length thermostated cell. The temperature was controlled by a circulating water-bath. The temperature changes were controlled by a programmable controller (Jasco Peltier) which increasing temperature of collagen from 20˚C to 45˚C.
Stability of collagen triple helix was calculated by using Spectra Manager Program and Statistica 8.
2.7. Bone Mineralization Assay
Osteoblasts were detected by staining for the presence of hydroxyapatite in the culture using Alizarin Red-S (AR-S), following fixation in cold 70% ethanol at −20˚C. At indicated osteoblast’s differentiation time points, the medium was removed and the culture was washed with PBS followed by fixation in ice-cold 70% ethanol for at least 1 hour. Subsequently, the ethanol was removed, and fixed cells were rinsed with de-ionized water, then stained with 40 mM AR-S (pH 4.2) for 10 minutes at room temperature. After staining the cells were further processed by rinsing them five times with water followed by 15 minutes wash with PBS to remove nonspecifically bound AR-S. The specimens were photographed and immediately processed for minerals content. Therefore, bound AR-S was solubilized with10% cetylpyridinium chloride (CPC) in 10 mM sodium phosphate, pH 7.0, for 15 minutes at 25˚C. Recovered AR-S was diluted 10-fold in 10% CPC solution, and the AR-S concentration was determined by measuring the absorbance at 562 nm wave length using an AR-S standard curve in the same solution [25,26]. The values were normalized per 1000 cells. The measurements were conducted in quadruplicates.
2.8. Isolation of BM-MSCs
Bone marrow derived mesenchymal stem cells (BMMSCs) were obtained from healthy patient’s father after written consent according to the protocol approved by the Jagiellonian University Bioethical Committee (KBET/108/B/2007). Bone marrow (150 ml) was harvested under anesthesia from the posterior superior iliac crest by multiple aspirations. Aspirates were collected in a Bone Marrow Collection Kit with Pre-Filter and Inline Filters (Baxter, USA) and further processed. Lightdensity mononuclear cells (MNC) were separated on a Lymphocyte Separation Medium (PAA Laboratories GmbH, Goetzis, Austria) by density gradient centrifugation at 400 g for 30 minutes at room temperature. MNC were washed twice with Dulbecco’s phosphate buffered saline (PAA Laboratories) and suspended in culture medium.
Culture of BM-MSCs
Bone marrow mononuclear cells were plated into vented 75 cm2 tissue culture flask (Sarstedt, Newton USA) with Dulbecco’s-Modified Eagles Medium (DMEM, SigmaAldrich Germany D5523) supplemented with 10% FBS (Stem Cell Technologies, Vancouver Canada 06472) and antibiotics (PAA Laboratories GmbH, Goetzis, Austria). Flasks were incubated at 37˚C in a humidified atmosphere containing 5% CO2 and after 7 days medium was replaced with a fresh one. Than the cells were cultured with a medium change every week until confluence. On reaching confluence, the adherent cells were detached using 0.25% trypsin and re-seeded at 75 cm2 flask (first passage). At the end of second passage when the cells reached confluence, they were trypsinized and checked for heamatopoietic contamination [27].
2.9. Flow Cytometry
MSC phenotype was analyzed with mouse antibodies specific for human CD45 and CD3 antigens (Becton Dickinson). Briefly, to 1 × 105 cells suspended in 100 µl of staining buffer (PBS, 2% FBS) 20 µl of mouse antibodies was added. Next, the cells were incubated in the dark for 30 minutes at 4˚C. Stained cells were washed, collected and analyzed using FACS Calibur cytometer (Becton Dickinson, USA).
2.10. Infusion of the MSC
Cells were harvested after the second passage in two parts in two weeks time. Each time cells were suspended in 10 ml of normal saline and passed through a filter before infusion. The patient received two 15- to 30-minutes intravenous infusions of MSCs 16 days apart. Target doses for the first and second infusions were 4.0 × 106 cells/kg of body weight and 3.9 × 106 cells/kg, respectively. Directly before infusion the patient received hydrocortisone. The girl was evaluated for toxicity (physical examination, complete blood count, clinical chemistry panel-BUN, glucose, AST, ALT, ALKP, urine analysis, before, and 24 hours after, each infusion. She was monitored at the bedside during, and for 6 to 8 hours after, each infusion. Nine days after first infusion she received a course of the Vancomycine because of positive blood culture for Staph. Epidermidis. During the bacteremia period she was stable and in a good general status. The period of two weeks after the second infusion was uneventful. Then she developed G(−) sepsis with Enterobacter cloacae treated with Meropenem and III generation cephalosporin antibiotics. She recovered from sepsis after two weeks and her general status was quite good.
Three months after MSC transplantation a bone marrow biopsy (for assessment the chimerism and cellular differentiation) and a skin biopsy (for evaluation the stability of collagen triple helix) were performed. Another blood sample was taken to molecular analysis of COL1A1 and COL1A2 genes.
The patient was finally discharged from hospital at the age of four months. She has had regular appointments in a genetic out-patient clinic (at the beginning every month, now every two months). Each visit consists of: history taking, physical examination, anthropometric measurements and collecting blood sample to assess the level of serum bone turn over markers.
3. RESULTS
3.1. Cells Expansion
Expansion of the cells resulted in harvesting 4 × 106 cells and 3.9 × 106 cells. Two infusions with collectively of 7.9 × 106 MSC were performed. Expanded cells contained 7.3% of hematopoietic contamination, but did not contain lymphocyte contamination (Figure 1).
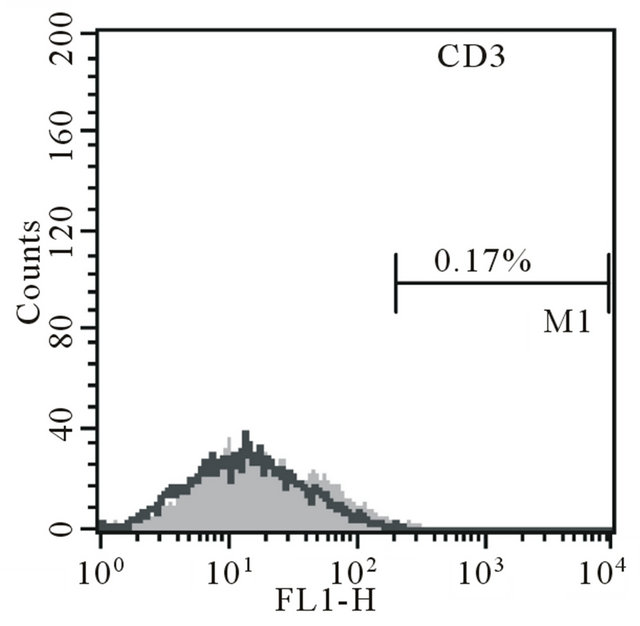
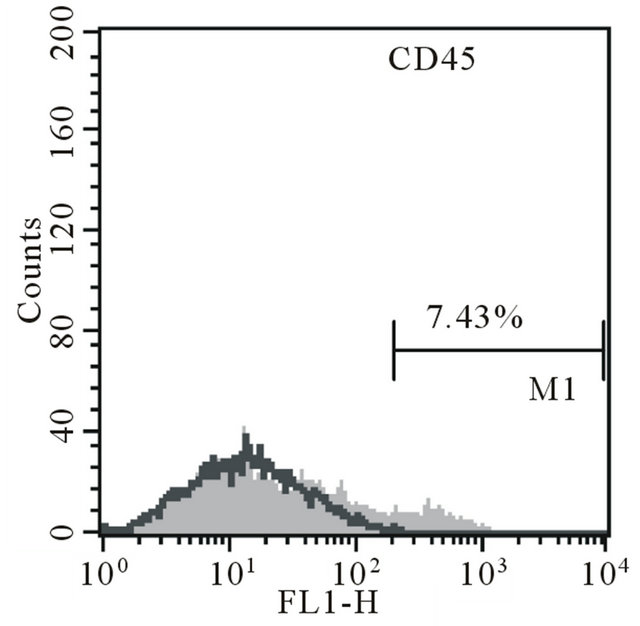
Figure 1. Majka M. et al. Cell therapy of a patient with type III osteogenesis imperfecta...
3.2. Patient Follow-Up
Patients follow up was performed monthly. Physical examination of the patient revealed short stature, severe lower limb deformities, relative macrocephaly, triangular face, blue sclera and dentinogenesis imperfecta. The antropometric measurements show extremely retarded growth (Table 2). The child’s psychomotoric development was not synchronic—motoric development is retarded, while the speech development and cognitive functions are adequate to the chronological age. The patient is under the constant care of physiotherapist, orthopedic surgeon, psychologist and dentist. An objective audiology (ABR, AOE) examination was not possible to perform so far. Nevertheless, there is no signs of hearing loss in our patient.
The patient has had six bone fractures since her birth of which five were identified during the performance of diagnostic bone marrow biopsy after the MSC infusion. During the afterwards follow up over the period of two years two new fractures were diagnosed. The results of serum bone markers levels did not revealed any clinically significant tendencies.
Stability of collagen triple helix assessed in skin fibroblasts taken prior to bone marrow MSC transplantation showed temperature of 2˚C lower than normal. Stability of collagen triple helix assessed in skin fibroblasts taken after the MSC transplantation returned to the normal temperature range (Figure 2).
The molecular analysis of the gene COL1A1 performed prior to and following the MSC transplantation did not reveal any mutation. A missense mutation
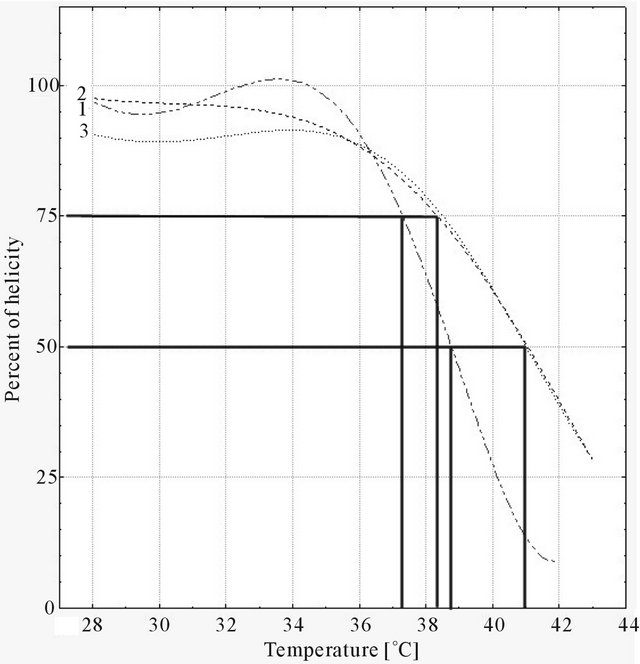
Figure 2. Majka M. et al. Cell therapy of a patient with type III osteogenesis imperfecta...
G23569A changing glycine at position 517 to aspartate (p.Gly517Asp) was detected in the exon 26 of COL1A2 gene before and after the MSC transplantation. This particular mutation was previously identified in one patient with type III osteogenesis imperfecta reported to Osteogenesis Imperfecta Variant Database.
3.3. Bone Mineralization
Data on differentiation of bone marrow cells to osteoblasts measured indirectly as mineral deposition are inconclusive. Although the bone marrow MSC collected following transplantation could be successfully expanded during process of differentiation their number decreased with time. The experiments were repeated three times in quadruplicates and none of them was successful. The control sample in this experiment was established culture of stem cells purified from human normal placenta. The cells grew accordingly during differentiation and revealed increasing mineral deposition expressed as amount of calcium ion bound to AS-R (Figure 3).
3.4. Chimerism
Genetic profile of transplanted patients were studied using AmpFlSTR SGM which recognized 11 STR loci. Only DNA of patient was detected indicating low engraftment of donor MSC.
4. DISCUSSION
Type III OI is known as progressive disease leading to serious skeletal deformations and severe handicap. Its presentation at birth is phenotypically similar to the milder end of the spectrum of type II disease. Although this form of the disorder is usually not life threatening, some children with type III OI die in infancy because of respiratory distress, whereas others die in childhood due to pneumonia, pulmonale or trauma. For those, who survive, there is progressive deformity of long bones and spine, which in combination with compression of verte brae, contributes to marked short stature. Most of the patients with type III OI are severely physically handicapped and have traditionally required multiple orthopedic procedures and wheelchairs for mobility.
In fact, there is no effective treatment for the patients with OI so far [27-30]. That is why, it seems to be of crucial importance to continue research studies investigating the role of new methods of therapy in patients with this disease. A development of new treatment could improve the prognosis and reduce unnecessary exposure to potentially painful therapies.
We conducted a study examining the role of cell therapy in patients with type II and III OI. Till now, we enrolled one trial subject. MSC were administered
Table 2. Sequences of the primers used for fragment amplification and for sequencing of the COL1A2 human gene.
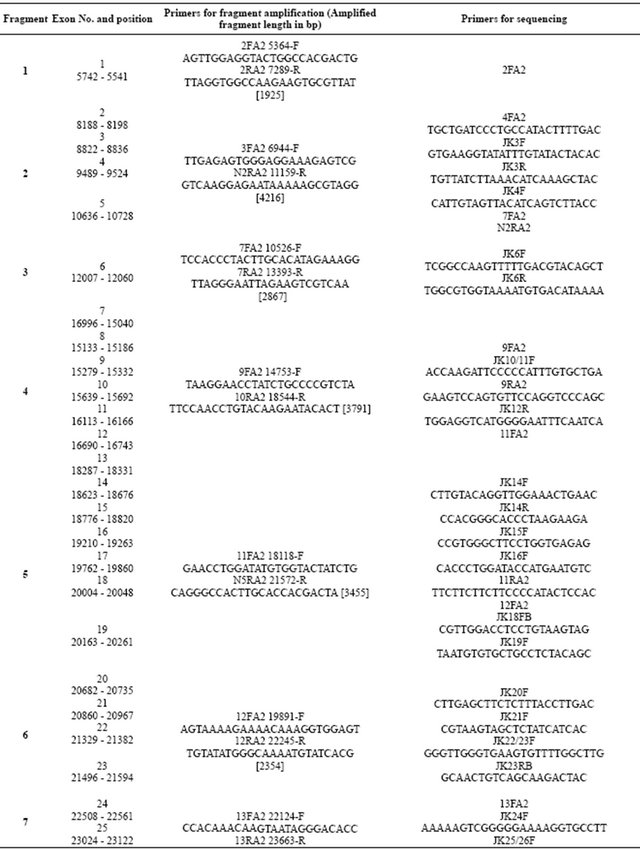
Continued

Continued

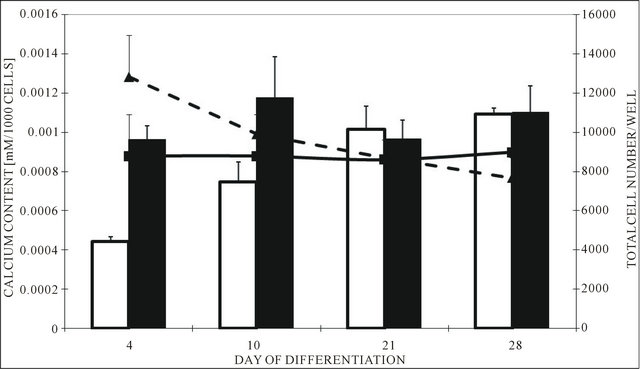
Figure 3. Majka M. et al. Cell therapy of a patient with type III osteogenesis imperfecta...
Intravenously. The most significant observation in the present study is the association between the MSC transplantation and the improvement of stability of collagen triple helix, and a decrease in a number of fractures observed in our patient. Stability of collagen triple helix after the MSC transplantation was normal, and there was only two bone fracture noted two years after the bone marrow stem cells transplantation. To validate this finding we measured serum bone markers levels, but they did not revealed any observable tendency. Nevertheless, during the first 2 years of life, the child had significant growth deficiency. Anyway, we can speculate that the cell therapy in patient with type III osteogenesis imperfecta looks promising. However, further evaluation is necessary.
Failure in detecting a chimerism in the patient could be a result that the threshold of the detection method used is 5%, thus, presence of donor cells could not be excluded if the cell engraftment fell under 5% and the donor DNA was not detected.
In summary, the stability of patient’s collagen triple helix assessed in skin fibroblasts taken after the mesenchymal cells transplantation was within a normal temperature range. The girl has had only two bone fractures two years after the transplantation. No cases of rejection reaction when the father’s MSCs are injected into the girl’s body were previously reported from other trials with the use of purified cells [17,18]. Therefore, in this study the graft versus host disease symptoms also were not observed. In conclusion, although, the assay of in vitro osteogenesis by minerals deposition was inconclusive, altogether the results indicate that they might be considered as an objective indicators of partial success of the cell therapy. It is very important for further clinical trials of cell therapy in the OI because no reports are available on remission of this disease, especially of its most severe type III.
5. ACKNOWLEDGEMENTS
The work was in part financed from Institutional grant KNW-1-010/ P/1/0 and KNW-1-007/P/2/0 awarded to ALS. Some equipment used to conduct the research was co-financed by the EU funds (European Regional Development Fund) within the Sectoral Operational Program “Increase of Economic Competitiveness”.
No. WKP1/1.4/3/2/2005/103/223/565/2007/U and Silesian BioFarma Center for Biotechnology, Bioengineering and Bioinformatics, Project no POIG.02.01.00-00-166/08, THE OPERATIONAL PROGRAMME INNOVATIVE ECONOMY FOR 2007-2013, Priority Axis 2. R & D Infrastructure.
![]()
![]()
REFERENCES
- Rauch, F. and Glorieux, F.H. (2004) Osteogenesis imperfecta. Lancet, 363, 1377-1385. doi:10.1016/S0140-6736(04)16051-0
- Plotkin, H. (2004) Syndromes with congenital brittle bones. BMC Pediatrics, 4, 16. doi:10.1186/1471-2431-4-16
- Glorieux, F.H. (2008) Osteogenesis imperfecta. Best Practice & Research Clinical Rheumatology, 22, 85-100. doi:10.1016/j.berh.2007.12.012
- Van Dijk, F.S., Pals, G., Van Rijn, R.R., Nikkels, P.G. and Cobben, J.M. (2010) Classification of osteogenesis imperfecta revisited. European Journal of Medical Genetics, 53, 1-5. doi:10.1016/j.ejmg.2009.10.007
- Hackley, L. and Merritt, L. (2008) Osteogenesis imperfecta in the neonate. Advances in Neonatal Care, 8, 21-30. doi:10.1097/01.ANC.0000311013.71510.41
- Morgan, J.A. and Marcus, P.S. (2010) Prenatal diagnosis and management of intrauterine fracture. Obstetrical & Gynecological Survey, 65, 249-259. doi:10.1097/OGX.0b013e3181dbc50b
- Tedeschi, E., Antoniazzi, F., Venturi, G., Zamboni, G. and Tatò, L. (2006) Osteogenesis imperfecta and its molecular diagnosis by determination of mutations of type I collagen genes. Pediatric Endocrinology Reviews, 4, 40- 46.
- Burnei, G., Vlad, C., Georgescu, I. and Gavriliu, T.S. (2008) Osteogenesis imperfecta: Diagnosis and treatment. Journal of the American Academy of Orthopaedic Surgeons, 16, 356-366.
- Basel, D. and Steiner, R.D. (2009) Osteogenesis imperfecta: Recent findings shed new light on this once well-understood condition. Genetics in Medicine, 11, 375-385. doi:10.1097/GIM.0b013e3181a1ff7b
- Marini, J.C., Cabral, W.A., Barnes, A.M., Chang and W. (2007) Components of the collagen prolyl 3-hydroxy-lation complex are crucial for normal bone development. Cell Cycle, 6, 1675-1681. doi:10.4161/cc.6.14.4474
- Tarnowski, M. and Sieroń, A.L. (2008) Osteogenesis imperfecta-etiology, characteristics, current and future treatment. Wiadomosci Lekarskie, 61, 166-172.
- Dent, C.E. and Davies, I.J. (1980) Calcium metabolism in bone disease: Effects of treatment with microcrystalline calcium hydroxyapatite compound and dihydrotachysterol. Journal of the Royal Society of Medicine, 73, 780- 785.
- Bachrach, L.K. and Ward, L.M. (2009) Clinical review 1: Bisphosphonate use in childhood osteoporosis. The Journal of Clinical Endocrinology & Metabolism, 94, 400- 409. doi:10.1210/jc.2008-1531
- Russell, R.G. (2007) Bisphosphonates: Mode of action and pharmacology. Pediatrics, 119, 150-162. doi:10.1542/peds.2006-2023H
- Benhamou, C.L. (2007) Effects of osteoporosis medications on bone quality. Joint Bone Spine, 74, 39-47. doi:10.1016/j.jbspin.2006.06.004
- Rauch, F. and Glorieux, F.H. (2005) Bisphosphonate treatment in osteogenesis imperfecta: Which drug, for whom, for how long? Annals of Medicine, 37, 295-302. doi:10.1080/07853890510007386
- Horwitz, E.M., Prockop, D.J., Fitzpatrick, L.A., et al., (1999) Transplantability and therapeutic effects of bone marrow-derived mesenchymal cells in children with osteogenesis imperfecta. Nature Medicine, 5, 309-313. doi:10.1038/6529
- Horwitz, E.M., Prockop, D.J., Gordon, P.L., et al., (2001) Clinical responses to bone marrow transplantation in children with severe osteogenesis imperfecta. Blood, 97, 1227-1231. doi:10.1182/blood.V97.5.1227
- Pochampally, R.R., Horwitz, E.M., DiGirolamo, C.M., Stokes, D.S. and Prockop, D.J. (2005) Correction of a mineralization defect by overexpression of a wild-type cDNA for COL1A1 in marrow stromal cells (MSCs) from a patient with osteogenesis imperfecta: A strategy for rescuing mutations that produce dominant-negative protein defects. Gene Therapy, 12, 1119-1125. doi:10.1038/sj.gt.3302514
- Niyibizi, C. and Li, F. (2009) Potential implications of cell therapy for osteogenesis imperfecta. International Journal of Clinical Rheumatology, 4, 57-66.
- Sillence, D.O., Senn, A. and Danks, D.M. (1979) Genetic heterogeneity in osteogenesis imperfecta. Journal of Medical Genetics, 16, 101-116. doi:10.1136/jmg.16.2.101
- Witecka, J., Auguściak-Duma, A.M., Kruczek, A., et al., (2008) Two novel COL1A1 mutations in patients with osteogenesis imperfecta (OI) affect the stability of the collagen type I triple-helix. Journal of Applied Genetics, 49, 283-295. doi:10.1007/BF03195625
- Fertala, A., Sieron, A.L., Ganguly, A., et al. (1994) Synthesis of recombinant human procollagen II in a stably transfected tumor cell line (HT-1080). Biochemical Journal, 298, 31-37.
- Arnold. W.V., Fertala, A., Sieron, A.L., et al. (1998) Recombinant procollagen II: Deletion of D period segments identifies sequences that are required for helix stabilization and generates a temperature-sensitive N-proteinase cleavage site. The Journal of Biological Chemistry, 273, 31822-31828. doi:10.1074/jbc.273.48.31822
- Stanford, C.M., Jacobson, P.A., Eanes, E.D., Lembke, L.A. and Midura, R.J. (1995) Rapidly forming apatitic mineral in an osteoblastic cell line (UMR 106-01 BSP). The Journal of Biological Chemistry, 270, 9420-9428. doi:10.1074/jbc.270.16.9420
- Gregory, C.A., Gunn, W.G., Peister, A. and Prockop, D.J. (2004) An Alizarin red-based assay of mineralization by adherent cells in culture: comparison with cetylpyridinium chloride extraction. Analytical Biochemistry, 329, 77-84. doi:10.1016/j.ab.2004.02.002
- Marini, J.C., Letocha, A.D. and Chernoff, E.J. (2005) Osteogenesis imperfecta. In: Cassidy, S.B and Allanson, J.E., Eds., Management of genetic syndromes. Published Online: 14 JAN 2005. http://onlinelibrary.wiley.com/doi/10.1002/0471695998.mgs034/abstract
- Esposito, P. and Plotkin, H. (2008) Surgical treatment of osteogenesis imperfecta: Current concepts. Current Opinion in Pediatrics, 20, 52-57. doi:10.1097/MOP.0b013e3282f35f03
- Marini, J. (2010) Osteogenesis imperfecta. In: Diseases of bone and calcium metabolism. South Dartmouth in US: Mdtext.com.inc. http://www.endotext.org/parathyroid/parathyroid17/parathyroidframe17.htm
- Chen, H. (2006) Osteogenesis imperfecta. In: Chen, H., Ed., Atlas of genetic diagnosis and counselling, Humana Press Inc., New Jersey, 762-772.
NOTES
*No conflict of interest exists for each author.
#Corresponding authors.