Open Journal of Internal Medicine
Vol.2 No.3(2012), Article ID:22962,3 pages DOI:10.4236/ojim.2012.23029
Brugada syndrome: A new mutation found in Norway*
![]()
1Medical Department, Ålesund Hospital, Ålesund, Norway
2Department of Cardiology, St. Olavs Hospital, Trondheim, Norway
3Norwegian University of Science and Tehcnology (NTNU), Trondheim, Norway
4Medical Genetics Laboratory, Oslo University Hospital Rikshospitalet, Oslo, Norway
Email: #eva-lill.rice@stolav.no
Received 12 March 2012; revised 12 April 2012; accepted 20 April 2012
Keywords: Brugada Syndrome; SCN5A; Mutation
ABSTRACT
Brugada syndrome is increasingly recognized as a clinical syndrome world wide. We report a case with a new SCN5A mutation. As the awareness of this disease entity increases more cases will probably be found.
1. INTRODUCTION
Brugada syndrome was first described as a clinical disease entity in 1992 and is predominantly seen in Asia. To our knowledge, only two cases have previously been diagnosed in Norway [1,2].
Brugada syndrome is a genetically inherited disease caused by dysfunction of myocardial sodium channels, known as a channelopathy. The dysfunction leads to abnormal sodium flux and thereby disturbances in the cardiomyocyte action potensial. Due to the nature of the underlying dysfunction, ECG changes will vary over time.
Three subtypes of repolarization patterns in ECG have been identified:
Type 1: Coved-shaped ST elevation in right precordial leads with J wave or ST elevation of at least 2 mm at its peak followed by a negative T wave with little or no isoelectric separation in more than one right precordial lead (V1-3);
Type 2: ST segment elevation followed by a positive or biphasic T wave that result in a saddle back configuration;
Type 3: Right precordial ST elevation of less than or equal to 1 mm with of saddle back type or coved type morphology.
Diagnostic criteria for Brugada syndrome have been published and defined in consensus reports published in 2002 and 2005 [3] as reviewed in the discussion part of this paper.
The syndrome can cause ventricular tachycardia (VT) and sudden death. It is also associated with supraventricular arrhythmias.
Treatment is aimed at prevention of arrythmias, evaluation of the need for implantation of an ICD, as well as genetic counselling [4,5].
2. CASE PRESENTATION
A 19 year old male was admitted to our clinic. He was unconscious upon arrival, Glasgow Coma Score of 3, improving to 15 over the next 4 hours. The patient was pyretic but otherwise stable. A suspected intake of amitriptyline was later disproven. Prehospital treatment had included resuscitation due to circulatory collapse and a 50 J DC cardioversion, reported to terminate an observed supraventricular tachycardia (SVT). On admission, he had sinus rhythm, but his ECG was abnormal and the tracings were variable (Figure 1). As his symptoms receded, the patient requested to be discharged. He was discharged but with a cardiologic follow up appointment to allow further investigations. However, he was readmitted a few weeks later due to increase palpitations, particularly during exercise, as well as episodes of syncope. A review of his ECGs by experienced cardiologists was warranted.
His presenting ECGs on the first admission showed sinus rhythm, incomplete right bundle branch block (RBBB) with coved ST segment elevations coexisting with an inverted T wave in V2, consistent with Brugada type 1 changes.
On readmission, ECG tracings revealed atrial flutter.
Repeated exercise ECGs induced a wide complex tachycardia that could represent SVT with bundle branch block pattern or a ventricular tachycardia (VT). There was no recorded VT in his history, and his syncope could be explained by either a VT or a 1:1 conducted atrial flutter. Repeated exercise ECG with simultaneous oesophageal lead recording revealed a 1:1 conducted atrial flutter with right bundle branch block.
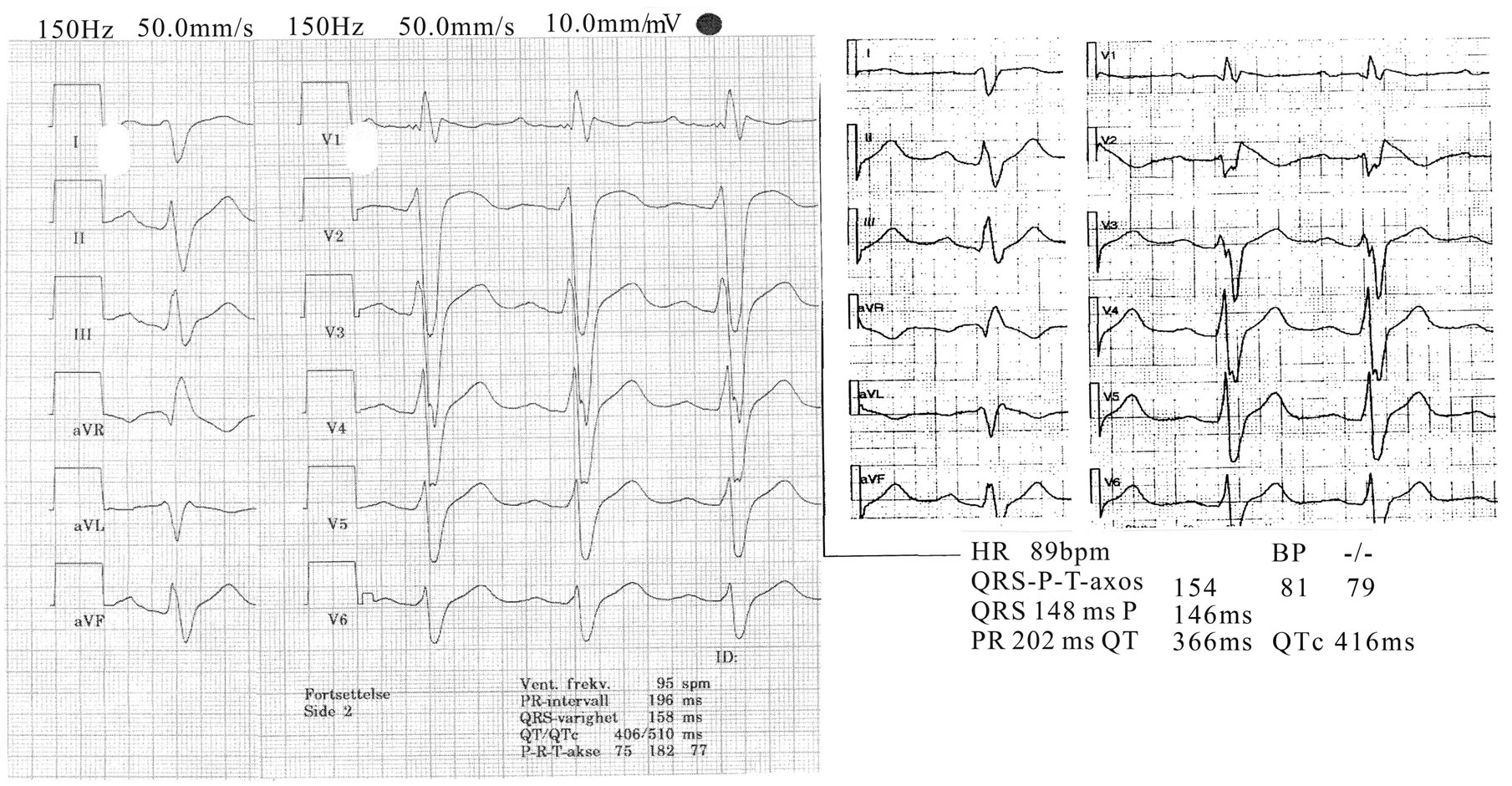
Figure 1. Standard 12 lead ECG of patient on 2 different occasions showing Brugada type 1 changes.
Our patient was referred to electrophysiological mapping with findings of a clockwise isthmus dependent atrial flutter which was successfully ablated. HV time was found to be pathologically increased at 85 ms both before and after ablation. Programmed ventricular stimulation at paced stimulation of 600 ms and 2 stimulations with S2-S3 at 250 ms induced a nonsustained regular VT with left bundle branch block pattern at 300 beats/min. Repeated stimulation induced a ventricular fibrillation with loss of consciousness, terminated by cardioversion.
A flecainide test was performed: After 40 mg administered intravenously, both a 30% widening of QRS width and concurrent coved ST segment elevation in V2 were seen.
Due to the findings of type 1 Brugada changes in the ECG, prolonged HV time, history of palpitations and syncope, he was diagnosed with Brugada syndrome. Because of a history of circulatory collapse requiring resuscitation, the patient received an ICD.
The patient’s parents were a native Norwegian and Singaporean, respectively. There was limited knowledge of his family history other than a possible sudden death during childhood in one of his Singaporean relatives.
Permission to perform a genetic analysis for SCN5A mutation was obtained. The results revealed a heterozygote C896Y (TGT > TAT) mutation in exon 16, a mutation not previously known to cause Brugada syndrome [6]. Analysis with PolyPhen prediction software indicated that this mutation was likely to cause abnormal function of myocardial sodium channels, leading to Brugada syndrome.
3. DISCUSSION
Brugada syndrome is a genetically inherited channelopathy with an autosomal dominant mode of transmission. Mutations in the SCN5A gene encoding the α subunit of the sodium channel has been found to cause a loss of function. Over 100 mutations have been found so far. It is known that mutation in SCN5A is linked to overlapping syndromes. Also, mutations in other genes have been reported in Brugada syndrome patients [4,5].
The diagnosis of Brugada syndrome is clinical. It is based on typical type 1 ECG findings, either spontaneous or on provocation, in combination with one of the following: Documented VF, polymorphic VT, family history of sudden cardiac death at age < 45, coved type ECG in family member, inducibility of VT with programmed stimulation, syncope or nocturnal agonal respiration attributed to self-terminated VT or VF.
Several provoking factors for type 1 changes in Brugada patients are known. These include fever, which our patient presented with, and certain drugs including amitriptyline, which our patient had been prescribed previously.
4. CONCLUSION
In our patient ECG recordings at rest, clinical history, electrophysiological testing and flecainide testing all support the diagnosis of Brugada syndrome, and a new SCN5A mutation was found.
REFERENCES
- Graven, T., Loennechen, J.P. and Leren, T.P. (2008) Brugadas syndrom. Tidsskr Nor Legeforen, 128, 2828- 2831.
- Holt, E. (2010) Brugadas syndrom. Hjerteforum, 23, 31- 38.
- Antzelevitch, C., Brugada, P., Borggrefe, M., et al. (2005) Brugada syndrome: Report of the second consensus conference: Endorsed by the Heart Rythm Society and the European Heart Rythm Association. Circulation, 111, 659-670. doi:10.1161/01.CIR.0000152479.54298.51
- Vohra, J. (2011) Diagnosis and management of Brugada syndrome. Heart, Lung and Circulation, 20, 751-756. doi:10.1016/j.hlc.2011.07.014
- Benito, B., Brugada, R., Brugada, J. and Brugada, P. (2008) Brugada syndrome. Progress in Cardiovascular Diseases, 51, 1-22. doi:10.1016/j.pcad.2008.05.002
- Andavan, G.B.S and Lemmens-Gruber, R. (2011) Voltage-gated sodium channels: Mutations, channelopathies and targets. Current Medicinal Chemistry, 18, 377-397. doi:10.2174/092986711794839133
NOTES
*Conflict of interest: None declared. The patient has agreed to the publication of his case history.
#Corresponding author.