Open Journal of Statistics
Vol.08 No.04(2018), Article ID:86831,10 pages
10.4236/ojs.2018.84047
Haplotype Frequency Comparison for Case-Parents Data
Caixia Li1, Peixing Li1,2*
1School of Mathematics, Sun Yat-sen University, Guangzhou, China
2Guangdong Province Key Laboratory of Computational Science, Sun Yat-sen University, Guangzhou, China

Copyright © 2018 by authors and Scientific Research Publishing Inc.
This work is licensed under the Creative Commons Attribution International License (CC BY 4.0).
http://creativecommons.org/licenses/by/4.0/



Received: July 25, 2018; Accepted: August 20, 2018; Published: August 23, 2018
ABSTRACT
For case-parents data, the information from offspring can be used to reduce the uncertainty of parents’ haplotype. In this article we develop likelihood ratio test to compare haplotype frequencies in transmitted and non-transmitted group. The maximum likelihood estimate of the haplotype frequencies for the family data is obtained via expectation-maximization (EM) algorithm. Our proposed method can handle the uncertainty of haplotypes and missing data. The simulations show that the method is more powerful to test association between haplotype and traits than TRANSMIT. We also demonstrated the method to detect the association between Megsin gene and immunoglobulin A nephropathy.
Keywords:
Haplotype, Transmission Disequilibrium Test, Likelihood Ratio Test, Expectation-Maximization Algorithm

1. Introduction
In association studies, to avoid false positive results caused by population stratification, family-based tests of association are often used for fine mapping of a disease susceptibility locus. Transmission disequilibrium test (TDT) proposed by Spielman is an association test for case-parents triad data [1] [2] . The classic TDT is McNemar test on a 2-way transmission/non-transmission table.
Multiple linked markers can provide more polymorphism information than single marker (especially SNPs). However, haplotype phase is often uncertain for multi-locus genotype. There may be several haplotype pairs compatible with observed genotype. Many haplotype reconstruction algorithms are developed, e.g. expectation-maximization (EM) algorithm [3] , pseudo MCMC [4] , Bayesian haplotype inference [5] for population data of unrelated individuals.
TDT has been extended for tightly linked marker loci. Zhao et al. (2000) [6] performed TDT via two steps: constructing the underlying haplotype first, and then constructing the 2-way transmission/non-transmission table in TDT by assigning a weight to each possible phase. Based on conditional likelihood, Clayton (1999) proposed a score test with program TRANSMIT [7] . The TDT-type methods were widely used in medical studies and GWAS studies [8] [9] .
In this paper, based on full likelihood, we proposed a likelihood ratio test integrating haplotype construction for case-parent or case-parents data.
2. Method
The key idea is from classical case-control association study. Association between trait and marker yields some allele or haplotypes are found more often in case group than control group. In case-parents data, the allele or haplotypes of the case are transmitted from the parents. And so, we can test whether a particular allele or haplotype exists more often in transmitted (case) than in non-transmitted (control).
For tightly linked marker loci, we treat haplotypes as extended alleles and use the transmission information to reduce the phase uncertainty.
Let H denote the number of haplotypes for l tightly linked loci. The set of all possible genotypes (haplotype pairs) is .
For a case-parents trios, the genotypes for father, mother and affected child are denoted by respectively. Let and denote haplotype frequency for transmitted and non-transmitted group, and denote relative risk for genotype i/j reference to H/H. Under multiplicative haplotype risk , where is relative risk for haplotype i reference to H. Under assumption of Hardy-Weinberg equilibrium, the probability of the father transmitted haplotype i and non-transmitted j, whereas the mother transmitted haplotype k and non-transmitted j to the child conditional on that the child is affected, is
(1)
where A means the child is affected, and
is regarded as frequency of haplotype i for disease population.
However, we only observed the genotype of each locus. The haplotype phase of the l tightly linked loci is often uncertainty, especial for missing parental genotype data. So there exist ambiguities to decide which haplotype is transmitted or not transmitted from the parent. Then
, (2)
where is the set of haplotype groups which haplotype pairs , , and are compatible with and , respectively. Here, missing parental genotype is allowable.
Suppose there are N case-parents trios, and then there are 2N parents in all. The genotypes for the r-th trios are . The log-likelihood
. (3)
It is difficult to find the maximum likelihood estimate (MLE) directly. We employ expectation-maximization (EM) algorithm to estimate the haplotype frequencies , by treating underlying haplotype pairs as “missing data”. The complete-data log-likelihood after adding missing data is
. (4)
We can show that the expected complete-data log-likelihood in E (expectation) step (see Appendix)
(5)
where
An iterative procedure can be used to find the MLE via EM algorithm. Given the current estimates , the estimates in the next step:
(6)
. Under the null hypothesis , we have
(7)
Let denote the Likelihood under the null hypothesis or . The likelihood ratio statistic
(8)
follows an asymptotic c2 distribution with H-1 degrees of freedom (df) when the null hypothesis is true.
According to the relationship between and
We can get
(9)
The maximum likelihood estimator (MLE) of the relative risk for haplotype i relative to haplotype H is therefore given as
(10)
3. Application
We apply our method to the published data which was used for family-based association analysis for immunoglobulin A nephropathy (IgAN) [10] . Two tightly linked loci C2093T and C2180T, located in the 3' untranslated region (UTR) in Megsin gene, were studied. This dataset contains 232 families with an affected child were entered into analysis (Table 1). There are missing data since the genotyping information of some parents are not available. Out of 232 nuclear families, only 125 (53.9%) families are complete case-parents trios for both loci, and 107 (46.1%) families are case-parent families for the locus C2093T or C2180T, which including 25 C2180T single parent families (SPF), 26 C2093T SPF, and 56 C2093T & C2180T SPF.
There exist 4 haplotypes for Megsin C2093T-C2180T, CC, CT, TC and TT, coded as 1, 2, 3 and 4. Give initial value and precision . The haplotype frequency estimates after the iterative procedure stops
The log-likelihood . For the null model, the haplotype frequency estimation
and log-likelihood . So likelihood ratio statistic
, df = 3, P = 0.0027. The results show the significant difference between F and F*, the transmitted haplotype frequencies

Table 1. 232 families entered into analysis.
SPF: single parent family.
and non-transmitted haplotype frequencies. is much higher than . That means haplotype 2 (2093C-2180T) is over-transmitted from parents to cases. In addition, reference to haplotype 4 (2093T-2180T), the estimated relative haplotype risk .
4. Simulations
In our simulations, two tightly linked single nucleotide polymorphism (SNP) marker genotype data for 100 case-parents trios are generated. In a similar way in Morris et al. (1997) [11] , samples are generated for simulation using the transmission model provided by Bickeboller et al. (1995) [12] .
where is penetrance for genotype , and is prevalence.
The frequencies of mutant disease allele D and normal allele d in a disease locus are denoted by
and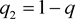 . Under Hardy-Weinberg Equilibrium, the population prevalence is therefore
. Under Hardy-Weinberg Equilibrium, the population prevalence is therefore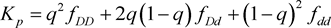 . We assume that there is no recombination between disease and marker locus. Linkage diseqilibrium parameter to detect association between disease locus and marker locus is defined as in Sham (1995) [13]
. We assume that there is no recombination between disease and marker locus. Linkage diseqilibrium parameter to detect association between disease locus and marker locus is defined as in Sham (1995) [13]

where 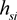 is frequency of disease-marker haplotype si. The
is frequency of disease-marker haplotype si. The  satisfies
satisfies
 and
and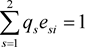 . All
. All  implies linkage
implies linkage
equilibrium. 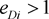 means marker haplotype i is positively associated with the disease, and
means marker haplotype i is positively associated with the disease, and 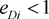 means marker haplotype i is negatively associated. In our simulations, the LD pattern is given as
means marker haplotype i is negatively associated. In our simulations, the LD pattern is given as

i.e. marker haplotype 1 is positively associated with the disease, and the other haplotypes are equally negatively associated, if there is association. In addition, the marker haplotypes are assumed to be equally frequent, and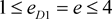 ,
,
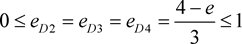 . Three classical genetic models, recessive, dominant
. Three classical genetic models, recessive, dominant
and common models, specified heredity modes are considered. These models are shown in Table 2.
For each genetic model, 5000 replicated samples were generated to evaluate the distribution of test statistics in the case of no association, i.e. e = 1. The quantile-quantile (QQ) plots for test statistic 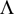 from 5000 samples under the null hypothesis for the three genetic models are showed in Figures 1(a)-(c). The
from 5000 samples under the null hypothesis for the three genetic models are showed in Figures 1(a)-(c). The
Table 2. Genetic models for simulation study.
 (a)
(a)
 (b)
(b)
 (c)
(c)
Figure 1. QQ plots for test statistic  from 5000 samples. Red: expected quantiles of distribution
from 5000 samples. Red: expected quantiles of distribution  versus observed quantiles; Green: the line y = x. (a) Recessive Model; (b) Dominant Model; (c) Common Model.
versus observed quantiles; Green: the line y = x. (a) Recessive Model; (b) Dominant Model; (c) Common Model.
plots show that the scatters of the observed quantiles and the expected quantiles of distribution  are very close to the line y = x.
are very close to the line y = x.
In addition, 1000 replicated samples were generated for statistical power analysis. The statistical significance level a = 0.05 or 0.01. The empirical power for our proposed method, comparison with TRANSMIT, are summarized in Table 3, under different association level e for three disease model.
Table 3. Comparisons of empirical power.
5. Discussion
Haplotype frequencies are usually estimated when haplotypes are reconstructed or linkage disequilibrium is tested. For tightly linked loci, the likelihood as a function of haplotype frequencies in transmitted and non-transmitted group was given for case-parents data. We estimate the MLEs of the haplotype frequencies via an EM algorithm. The results showed that haplotype frequencies could be estimated using a simple iterative procedure. The likelihood ratio test to compare haplotype frequencies in transmitted and non-transmitted group was used to detect association. When the information of parents is not available in the nuclear family, classical TDT is no longer suitable. However, as you can see in the application, missing parental genotypes are allowed in our method. In addition, when there are siblings available, the information can be used to reduce the uncertainty of phase, and the likelihood can be given similarly.
Under different simulated conditions where heredity mode, linkage disequilibrium coefficient are specified, 5000 and 1000 replicated samples were generated to evaluate the distribution of test statistics and statistical power respectively. Our method is more powerful than TRANSMIT.
Conflicts of Interest
The authors declare no conflicts of interest regarding the publication of this paper.
Cite this paper
Li, C.X. and Li, P.X. (2018) Haplotype Frequency Comparison for Case-Parents Data. Open Journal of Statistics, 8, 721-730. https://doi.org/10.4236/ojs.2018.84047
References
- 1. Spielman, R.S., McGinnis, R.E. and Ewens, W.J. (1993) Transmission Test for Linkage Disequilibrium: The Insulin Gene Region and Insulin-Dependent Diabetes Mellitus. American Journal of Human Genetics, 52, 506-516.
- 2. Spielman, R.S. and Ewens, W.J. (1996) The TDT and Other Family-Based Tests for Linkage Disequilibrium and Association. American Journal of Human Genetics, 59, 983-989.
- 3. Clayton, D.G. (1999) Transmission/Disequilibrium Tests for Extended Marker Haplotypes. American Journal of Human Genetics, 65, 1161-1169. https://doi.org/10.1086/302566
- 4. Stephens, M., Smith, N.J. and Donnelly, P. (2001) A New Statistical Method for Haplotype Reconstruction from Population Data. American Journal of Human Genetics, 68, 978-989. https://doi.org/10.1086/319501
- 5. Niu, T., Qin, Z.S., Xu, X. and Liu, J.S. (2002) Bayesian Haplotype Inference for Multiple Linked Single-Nucleotide Polymorphisms. American Journal of Human Genetics, 70, 157-169. https://doi.org/10.1086/338446
- 6. Zhao, H., Zhang, S., Merikangas, K.R., Trixler, M., Wildenauer, D.B., Sun, F., Kidd, K.K. (2000) Transmission/Disequilibrium Test Using Multiple Tightly Linked Markers. American Journal of Human Genetics, 67, 936-946. https://doi.org/10.1086/303073
- 7. Clayton, D.G. (1999) A Generalization of the Transmission/Disequilibrium Test for Uncertain-Haplotype Transmission. American Journal of Human Genetics, 65, 1170-1177. https://doi.org/10.1086/302577
- 8. Abdulkadir, M., Londono, D., Gordon, D., et al. (2017) Investigation of previously Implicated Genetic Variants in Chronic Tic Disorders: A Transmission Disequilibrium Test Approach. European Archives of Psychiatry and Clinical Neuroscience, 268, 1-16.
- 9. Wang, M., Ji, Z., Wang, S., et al. (2017) Mechanisms to Protect the Privacy of Families When Using the Transmission Disequilibrium Test in Genome-Wide Association Studies. Bioinformatics, 33,
- 10. Li, Y.J., Du, Y., Li, C.X., et al. (2004) Family-Based Association Study Showing That Immunoglobulin a Nephropathy Is Associated with the Polymorphisms 2093C and 2180T in the 3' Untranslated Region of Megsin Gene. Journal of the American Society of Nephrology, 15, 1739-1743. https://doi.org/10.1097/01.ASN.0000130858.00688.46
- 11. Morris, A.P., Whittaker, J.C. and Curnow, R.N. (1997) A Likelihood Ratio Test for Detecting Patterns of Disease-Marker Association. Annals of Human Genetics, 61, 335-350. https://doi.org/10.1017/S0003480097006349
- 12. Bickeboller, H. and Clerget-Darpoux, F. (1995) Statistical Properties of the Allelic and Genotypic Transmission/Disequilibrium Test for Multiallelic Markers. Genetic Epidemiology, 12, 865-870. https://doi.org/10.1002/gepi.1370120656
- 13. Sham, P.C. and Curtis, D. (1995) An Extended Transmission/Disequilibrium Test (TDT) for Multi-Allele Marker Loci. Annals of Human Genetics, 59, 323-336. https://doi.org/10.1111/j.1469-1809.1995.tb00751.x
Appendix (E Step in EM Algorithm)
Give the current estimate , the distribution of missing data
, the distribution of missing data

where

And then the expected complete-data log-likelihood
