International Journal of Organic Chemistry
Vol.3 No.1(2013), Article ID:29083,7 pages DOI:10.4236/ijoc.2013.31006
Synthesis and Characterization of Luteinizing Hormone-Releasing Hormone (LHRH)-Functionalized Mini-Dendrimers
1School of Chemistry and Molecular Biosciences, The University of Queensland, Brisbane, Australia
2School of Pharmacy, The University of Queensland, Brisbane, Australia
Email: *f.mansfeld@uq.edu.au
Received January 8, 2013; revised February 12, 2013; accepted February 23, 2013
Keywords: LHRH; Poly-Lysine; Dendrimer; Convergent Synthesis
ABSTRACT
Luteinizing hormone-releasing hormone (LHRH) is the key regulator of the hypothalamic-pituitary-gonadal (HPG) axis, which is responsible for the development and functioning of the reproductive system. Delivery of a continuous supply of LHRH agonists causes down-regulation of the LHRH receptors, resulting in a marked decrease in androgens in males and estrogens in females. LHRH analogues are widely used in the treatment of various diseases, including prostate and breast cancer, and reproductive disorders, such as infertility and precocious puberty. However, they require parenteral administration, and no oral formulations are currently available. We synthesized two types of LHRH mini-dendrimers using thioether ligation, aiming to enhance the stability and bioavailability of the peptide drug while maintaining its biologically active conformation. These two compounds include a poly-lysine core conjugated to either the C-terminus of LHRH or a D-amino acid in position 6 of the LHRH sequence. The synthesized dendrimers were analysed using dynamic light scattering, and showed particle sizes of 155 and 115 nm, respectively. The nanometer size, well-defined structure and water solubility of these dendritic analogues make them excellent candidates for further exploration in oral peptide drug delivery.
1. Introduction
Luteinizing hormone-releasing hormone (LHRH), also known as gonadotropin-releasing hormone (GnRH), is the central regulator of the hormones of the hypothalamic-pituitary-gonadal (HPG) axis, which controls and regulates reproductive function [1]. This decapeptide is released from neurons of the hypothalamus into the portal vascular system in synchronized pulses. It is then transported to the anterior pituitary where it activates its receptor, resulting in the production and secretion of the gonadotropins follicle-stimulating hormone (FSH) and luteinizing hormone (LH). FSH and LH are transported via the bloodstream to their receptors on the gonads and induce spermatogenesis and oogenesis as well as the production of sex hormones and gonadal peptide hormones [1-3]. Continuous supply of LHRH, as opposed to the naturally occurring pulsatile pattern, causes downregulation of the LHRH receptors, resulting in a marked decrease in androgens in males and estrogens in females. This has been exploited in the treatment of hormonesensitive cancers, such as prostate, ovarian and breast cancers, as well as reproductive disorders, including endometriosis and uterine fibroids [4-6].
Native LHRH has a short half-life of approximately 3 - 4 minutes in human blood, resulting from enzymatic degradation of the peptide, mainly at positions 6 and 10 of the LHRH sequence [7]. Since the amino acid sequence elucidation of LHRH (Figure 1) by Schally et al. in 1971 [8], thousands of LHRH analogues have been synthesized with the aim of increasing the stability and bioavailability of the peptide [7]. Substitution of the amino acids in positions 6 and 10 of the LHRH sequence was found to enhance the stability of the LHRH derivatives [6]. In addition, it was shown that the biologically active conformation of the LHRH peptide contains a β type II turn, which is stabilized by replacement of the glycine in position 6 with a D-amino acid [9,10].
All currently available LHRH agonist drugs require either subcutaneous or intramuscular injection and no oral formulation is available [3]. Although oral delivery of therapeutics is a convenient route of administration and associated with higher patient compliance, it is not
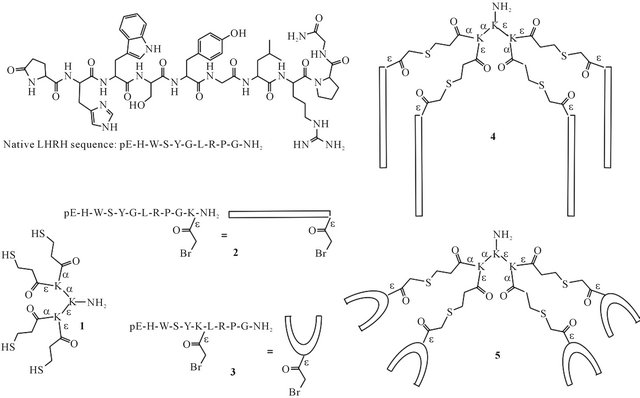
Figure 1. Structure and amino acid sequence of native LHRH. Structures of tetrathiol dendron core 1, bromoacetylated LHRH derivatives 2 and 3, and mini-dendrimeric LHRH derivatives 4 and 5.
easily achievable. The main reasons are the poor permeability of peptides across the intestinal epithelium and rapid enzymatic degradation in the environment of gastrointestinal (GI) tract [11-13]. Therefore, development of a potent water-soluble carrier system, which enhances the stability and bioavailability of the peptide drug while maintaining its biological activity, is essential for oral peptide drug delivery.
The potential application of dendrimers as drug delivery vectors has received much attention in the pharmaceutical field, particularly in the area of oral drug delivery, owing to unique features such as their well-defined structure, nanometer size, polyvalency, narrow polydispersity and structural flexibility [14,15]. Dendrimers have also shown higher stability against enzymatic degradation compared to other carriers such as liposomes or colloidal carriers [12]. Several types of dendrimers with different functionalities and architectures, mostly polyamidoamine (PAMAM)-based dendrimers and poly-lysine dendrimers, have been investigated [16-18]. However, the biodegradable polylysine dendrimers were found to exhibit lower toxicity than PAMAM based dendrimers, which accumulate in the liver and spleen after intravenous injection [19-21].
Here, we report a synthetic route to multivalent LHRH derivatives based on the poly-lysine peptide dendrimer, with the aim of creating stable, well-defined agonist candidates. LHRH was coupled to the poly-lysine core using thioether ligation, either through an amino acid added at the C-terminus, or a D-amino acid placed in position 6 of the peptide sequence (4 and 5, Figure 1). The synthesized mini-dendrimers were characterized using reversephase high performance liquid chromatography (RPHPLC), electrospray ionization mass spectrometry (ESIMS) and dynamic light scattering (DLS).
2. Material and Methods
2.1. Materials
Rink amide MBHA resin (100 - 200 mesh, 0.6 mmol/g) was purchased from Novabiochem. Fmoc-protected amino acids with acid-labile side chain protecting groups, such as tBu (Ser and Tyr), Boc (Trp), Trt (His) and Pbf (Arg), base-labile side chain protecting protected groups, such as Fmoc (Lys), Dde (D-Lys) or iv-Dde (Lys), and unprotected amino acids such as Gly, Leu, Pro and pyroglutamic acid (pGlu), were purchased from Mimotopes (Clayton, Vic, Australia). S-trityl-3-mercaptopropionic acid (Trt-Mpa) was purchased from Mimotopes (Clayton, Vic, Australia). N,N-dimethylformamide (DMF), trifluoroacetic acid (TFA), piperidine and N,N-diisopropylethylamine (DIPEA) were purchased from Merck (Kilsyth, Vic, Australia); 1-[bis(dimethylamino)methylene]-1H-1,2,3-triazolo [4,5-b]pyridinium 3-oxid hexafluorophosphate (HATU) was purchased from Mimotopes (Clayton, Vic, Australia). HPLC grade acetonitrile (MeCN) was obtained from RCI Labscan Ltd. (Bangkok, Thailand). All other chemicals were purchased from Sigma Aldrich (Castle Hill, NSW, Australia).
Microwave assisted Fmoc chemistry solid-phase peptide synthesis (SPPS) was accomplished by using a SPPS mode CEM Discovery reactor (CME Corporation, Matthews, NC, USA). Analytical reverse-phase high performance liquid chromatography (RP-HPLC) was performed on a Shimadzu instrument (Kyoto, Japan), equipped with a SIL-20AC HT autosampler, LC-20AB pumps with a flow rate of 1 mL/min and detection at 214 nm. Preparative RP-HPLC was performed by a Shimadzu system with a flow rate of 20 mL/min. Analytical separation of compounds was achieved using C4 (4.6 × 250 mm, 5 μm) or C18 (4.6 × 250 mm, 5 μm) Vydac columns, and for preparative RP-HPLC C4 (22 × 250 mm, 10 μm) or C18 (22 × 250 mm, 10 μm) Vydac columns were used. Solvent A (H2O, 0.1% TFA) and solvent B (MeCN/H2O 9:1, 0.1% TFA) were used for both analytical and preparative RP-HPLC. Electrospray ionization mass spectrometry (ESI-MS) was performed on a PE Sciex API3000 triple quadrupole instrument. Dynamic light scattering was accomplished using a Zetasizer (Malvern, England).
2.2. Peptide Synthesis and Purification
Synthesis of peptides was performed on Rink amide MBHA resin using microwave-assisted Fmoc-SPPS. Fmoc protected amino acids (4.2 eq.), activated with HATU (4 eq.) and DIPEA (6 eq.), were double-coupled to the resin after deprotection of Fmoc protecting group by treatment with 20% piperidine in DMF.
The core peptide (1) was also synthesized on Rink amide MBHA resin, by introducing Fmoc-Lys(Fmoc) as a branching unit, and functionalized with Trt-Mpa (8 eq., 2 eq. per branch of the tetravalent dendrone core) activated with HATU (8 eq.) and DIPEA (10 eq.). The bromoacetylated LHRH derivatives 2 and 3 were synthesized by introducing Fmoc-D-Lys(Dde)-OH in position 6 or addition of Fmoc-Lys(ivDde)-OH at the C-terminus of the LHRH sequence, respectively. Deprotection of the lysine side chain was accomplished with 2.5% hydrazine hydrate in DMF (4 × 20 min for Dde, 9 × 20 min for iv-Dde). Peptides 2 and 3 were subsequently functionalized by treatment with a mixture of bromoacetic acid (10 eq.) and N,N-diisopropylcarbodiimide (5 eq.).
Once assembly of the peptides was completed, the resin was washed with DMF and dichloromethane and dried under vacuum over night. Peptides were cleaved by treating the resin with TFA (94%), 1,2-ethanedithiol (2.5%), tri-isopropylsilane (2.5%) and water (1%) (1), or TFA (95%), tri-isopropylsilane (2.5%) and water (2.5%) (2 and 3) for three hours. Following precipitation with cold diethyl ether, the peptide was dissolved in MeCN/ H2O 1:1 with 0.1% TFA and lyophilized.
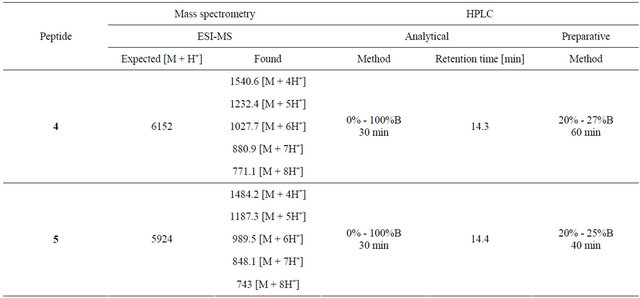
HPLC analysis and purification of all peptides was carried out using a Vydac C18 column and a gradient of solvents A and B specified in Table 1. Fractions collected from preparative HPLC were analyzed using electrospray ionization mass spectrometry (ESI-MS) and analytical HPLC and pure fractions were combined and lyophilized.
2.3. Peptide Ligation
Dendritic constructs 4 and 5 were synthesized from their pure precursors, the tetrathiol dendron core (1) and LHRH peptide derivatives 2 or 3, respectively. Compound 1 (2 μmol) and tris(2-carboxyethyl)phosphine (TCEP, 4 μmol) were dissolved in 0.4 ml MeCN and 0.6 mL aqueous sodium bicarbonate solution (20 mM, pH 7.5). Either peptide 2 or 3 (20 μmol) was dissolved in sodium bicarbonate solution (9 mL, 20 mM, pH 7.5), which had been degassed by bubbling argon through for 30 minutes. The solutions were mixed and the ligation reaction was monitored by analytical HPLC (gradient 15% - 30% solvent B over 30 minutes, Vydac C4 column) and ESI-MS. After 5 hours, the mixtures were injected onto a preparative HPLC column (C4) using the method shown in Table 2. Collected fractions were analysed by analytical HPLC and ESI-MS and pure fractions were
Table 1. Mass spectrometry and HPLC analysis of peptides 1-3.
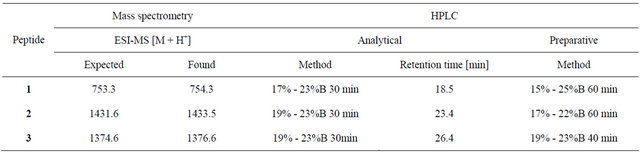
Table 2. Mass spectrometry and HPLC analysis of peptides 4 and 5.
combined and lyophilized.
2.4. Dynamic Light Scattering
Aggregation of the mini-dendrimeric LHRH derivatives was assessed by dynamic light scattering. Peptides were dissolved in water (200 μM) and each intensity graph was obtained by taking the average of twelve measurements
3. Results and Discussion
3.1. Peptide Desing and Synthesis
Poly-lysine dendrimers, pioneered by Tomalia et al. [22], are recognized as potent carriers for therapeutic compounds due to their well-defined structure, stability, nanometer size, narrow polydispersity and structural flexibility. They can be synthesized using the divergent approach, building the dendrimer from the core to the surface, or the convergent approach, building the dendrimer from the surface to the core from separately synthesized and purified precursors [14].
Initial attempts at generating our dendritic LHRH agonist candidates using either Boc or Fmoc solid phase peptide synthesis for the divergent approach were unsuccessful. The product obtained from the Boc synthesis was an insoluble gel, and although Fmoc chemistry gave the correct product, the yield was very poor and it was exceedingly difficult to separate from the numerous side products. Therefore, we decided to focus our synthetic efforts on using the convergent approach.
A wide variety of chemistries has been employed for convergent synthesis of polyvalent peptide species, leading to the formation of different types of bonds, including hydrazone, oxime, thioester and thioether bonds [23]. As the formation of thioethers from thiols and haloacetyl-functionalized compounds usually proceeds without difficulty and leads to stable products, we chose this approach. Thioether ligation, however, between a tetrabromoacetylated core and thiol-containing LHRH derivatives did not proceed to completion and only small amounts of the desired product were formed (data not shown).
The synthesis of dendritic constructs using thioether ligation between a thiol-functionalized dendron core and haloacetyl-functionalized peptides has several advantages over other ligation reactions [24]. Only a low amount of reducing agent is required for prevention of disulphide bond formation, minimizing the risk of haloacetyl degradation, resulting in better yields and faster ligation reaction rates [24].
Tetrathiol dendron core 1 and the LHRH derivatives 2 and 3 (Figure 1) were synthesized using Fmoc solid phase peptide synthesis. The bromoacetylated LHRH derivatives were synthesized using either Lys (ivDde) or D-Lys (ivDde) at the appropriate position in the amino acid sequence [25,26]. Once the peptides were fully assembled, the protecting group was removed from the lysine side chain and the resulting free amine was functionalized with a bromoacetyl group. Peptide 3 contains bromoacetyl-functionalized D-Lys in position 6. As discussed in the introduction, D-amino acids in position 6 stabilize a conformation important for interaction with the receptor. Additionally, ligating the LHRH derivative to the core through this position leaves the Nand the C-terminus free, both of which are involved in receptor binding [2]. Compound 2 contains a bromoacetylated Lys residue added at the C-terminus of the LHRH amino acid sequence. Ligation through a C-terminal amino acid may still result in active constructs, as activation of the receptor occurs exclusively through the N-terminal sequence [2].
3.2. Peptide Ligation
The thioether ligation between purified peptides 1 and 2, or 1 and 3, was carried out in the presence of TCEP to avoid disulphide formation of the thiol groups on 1. The use of only a low amount of TCEP (0.5 eq. per branch of the core peptide 1) and an excess of bromoacetyl peptides 2 or 3 (2.5 eq. per branch of the core peptide 1) limits the degradation of the bromoacetyl functionality [24]. This leads to a reaction mixture with very small amounts of undesired side products, as illustrated in Figure 2 for the reaction between 1 and 2. Additionally, the ligation proceeds rapidly, is near completion within one hour and was terminated after five hours when no further change was observed (Figure 2).
Upon completion of the ligation reaction, the mixtures were injected onto a preparative HPLC column and highly pure products 4 and 5 were obtained in good yields after a single purification (Figures 3 and 4).
3.3. Dynamic Light Scattering
The aggregation of peptides and the size of the resulting particles play a very important role in drug delivery. Their ability to cross biological barriers, such as the epithelium of the gastrointestinal tract, and the mechanisms accessible for this process, is highly dependent on the particle size [14]. Aggregates below 50 nm can pass the intestinal epithelium by paracellular transport, bigger particles are absorbed by endocytosis or the M cells of Peyer’s patches [27]. Therefore, to determine if aggregation of the mini-dendrimeric LHRH agonist candidates 4 and 5 occurs and how big the resulting particles are, we performed dynamic light scattering.
Average sizes of 115 nm with a polydispersity index (PDI) of 0.16 and 155 nm with a PDI of 0.25 were observed for 4 and 5 (Figure 5), respectively. Aggregates of 5 showed two distinct populations, with sizes of 1.2 nm and 139 nm, whereas solutions of 4 contain aggregates of only one size. Although the peptides coupled to the core consist of very similar sequences, the location of attachment clearly influences intermolecular interaction, resulting in assemblies of different sizes. If the aggregates stay intact during gastrointestinal transit, different uptake routes may be accessible to these two compounds.
4. Conclusion
Two types of dendritic LHRH agonist candidates were
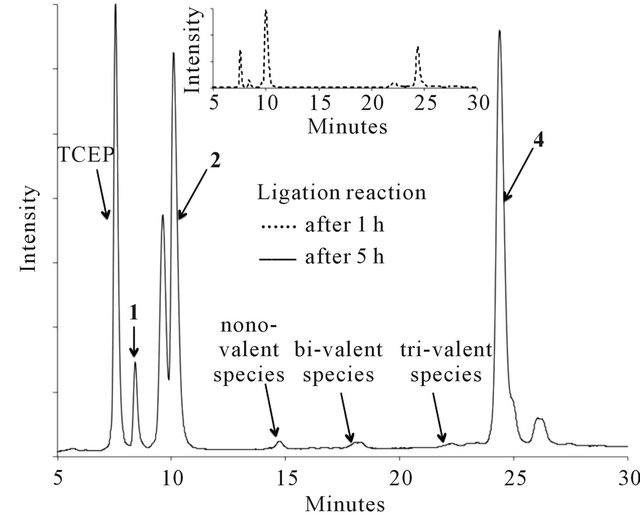
Figure 2. HPLC traces showing the development of the thioether ligation between 1 and 2 over 5 hours.
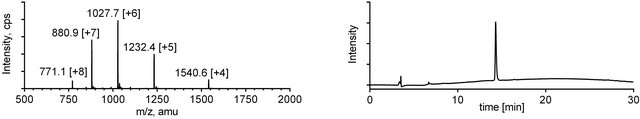
Figure 3. ESI-MS (left) and analytical RP-HPLC (right) traces for mini-dendrimer 4.
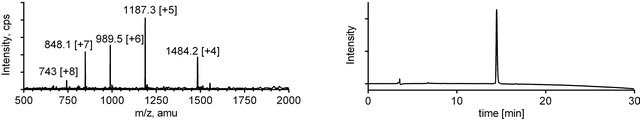
Figure 4. ESI-MS (left) and analytical RP-HPLC (right) traces for mini-dendrimer 5.
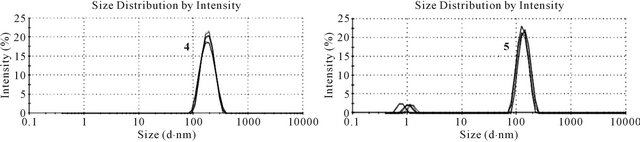
Figure 5. Dynamic light scattering of tetravalent LHRH agonist candidates 4 (left) and 5 (right). Six measurements of twelve runs were performed for each compound.
synthesized via thioether ligation. This technique allowed the generation of highly pure constructs with excellent yields. The mini-dendrimers were tested for aggregation using dynamic light scattering, showing average particle sizes of approximately 155 nm and 115 nm for compounds 4 and 5, respectively. The nanometer size, welldefined structure and water solubility of these dendritic analogues make them excellent candidates for further exploration in oral peptide drug delivery.
5. Acknowledgements
We are grateful for funding provided by the Australian Research Council (DP110100212). I.T. was supported by an Australian Research Council (ARC) Australian Professional Fellowship (DP110100212). P.M.M. was supported by a NHMRC postdoctoral training fellowship (569869).
REFERENCES
- S. V. Meethal and C. S. Atwood, “The Role of Hypothalamic-Pituitary-Gonadal Hormones in the Normal Structure and Functioning of the Brain,” Cellular and Molecular Life Sciences: CMLS, Vol. 62, No. 3, 2005, pp. 257-270. doi:10.1007/s00018-004-4381-3
- R. P. Millar, Z. L. Lu, A. J. Pawson, C. A. Flanagan, K. Morgan and S. R. Maudsley, “Gonadotropin-Releasing Hormone Receptors,” Endocrine Reviews, Vol. 25, No. 2, 2004, pp. 235-275. doi:10.1210/er.2003-0002
- D. A. Beyer, F. Amari, M, Thill, A. Schultze-Mosgau, S. Al-Hasani, K. Diedrich and G. Griesinger, “Emerging Gonadotropin-Releasing Hormone Agonists,” Expert Opinion on Emerging Drugs, Vol. 16, No. 2, 2011, pp. 323-340. doi:10.1517/14728214.2010.547472
- J. Sandow, “Clinical Applications of LHRH and Its Analogues,” Clinical Endocrinology, Vol. 18, No. 6, 1983, pp. 571-592.
- J. B. Engel and A. V. Schally, “Drug Insight: Clinical Use of Agonists and Antagonists of Luteinizing-HormoneReleasing Hormone,” Nature Clinical Practice Endocrinology & Metabolism, Vol. 3, No. 2, 2007, pp. 157-167. doi:10.1038/ncpendmet0399
- M. J. Karten and J. E. Rivier, “Gonadotropin-Releasing Hormone Analog Design. Structure-Function Studies toward the Development of Agonists and Antagonists: Rationale and Perspective,” Endocrine Reviews, Vol. 7, No. 1, 1986, pp. 44-66. doi:10.1210/edrv-7-1-44
- J. P. Moreau, P. Delavault and J. Blumberg, “Luteinizing Hormone-Releasing Hormone Agonists in the Treatment of Prostate Cancer: A Review of Their Discovery, Development, and Place in Therapy,” Clinical Therapeutics, Vol. 28, No. 10, 2006, pp. 1485-1508. doi:10.1016/j.clinthera.2006.10.018
- H. Matsuo, Y. Baba, R. M. G. Nair, A. Arimura and A. V. Schally, “Structure of the Porcine LHand FSH-Releasing Hormone. I. The Proposed Amino Acid Sequence,” Biochemical and Biophysical Research Communications, Vol. 43, No. 6, 1971, pp. 1334-1339. doi:10.1016/S0006-291X(71)80019-0
- C. A. Flanagan, R. P. Millar and N. Illing, “Advances in Understanding Gonadotrophin-Releasing Hormone Receptor Structure and Ligand Interactions,” Reviews of Reproduction, Vol. 2, No 2, 1997, pp. 113-120. doi:10.1530/ror.0.0020113
- G. V. Nikiforovich and G. R. Marshall, “Conformation-Function Relationships in LHRH Analogs,” International Journal of Peptide and Protein Research, Vol. 42, No. 2, 1993, pp. 181-193. doi:10.1111/j.1399-3011.1993.tb00495.x
- J. H. Hamman, G. M. Enslin and A. F. Kotzé, “Oral Delivery of Peptide Drugs: Barriers and Developments,” Biodrugs, Vol. 19, No. 3, 2005, pp. 165-177.
- M. Morishita and N. A. Peppas, “Is the Oral Route Possible for Peptide and Protein Drug Delivery?” Drug Discovery Today, Vol. 11, No. 19, 2006, pp. 905-910. doi:10.1016/j.drudis.2006.08.005
- H. Chen and R. Langer, “Oral Particulate Delivery: Status and Future Trends,” Advanced Drug Delivery Reviews, Vol. 34, No. 2, 1998, pp. 339-350. doi:10.1016/S0169-409X(98)00047-7
- B. K. Nanjwade, V. K. Nanjwade, H. M. Bechra, G. K. Derkar and F. V. Manvi, “Dendrimers: Emerging Polymers for Drug-Delivery Systems,” European Journal of Pharmaceutical Sciences, Vol. 38, No. 3, 2009, pp. 185- 196. doi:10.1016/j.ejps.2009.07.008
- V. V. Ranade, “Drug Delivery Systems,” CRC Press, Hoboken, 2011.
- A. T. Florence, T. Sakthivel and I. Toth, “Oral Uptake and Translocation of a Polylysine Dendrimer with a Lipid Surface,” Journal of Controlled Release, Vol. 65, No. 1, 2000, pp. 253-259. doi:10.1016/S0168-3659(99)00237-0
- Y. Cheng, Z. Xu, M. Ma and T. Xu, “Dendrimers as Drug Carriers: Applications in Different Routes of Drug Administration,” Journal of Pharmaceutical Sciences, Vol. 97, No. 1, 2007, pp. 123-143. doi:10.1002/jps.21079
- R. Esfand and D. A. Tomalia, “Poly (Amidoamine) (PAMAM) Dendrimers: From Biomimicry to Drug Delivery and Biomedical Applications,” Drug Discovery Today, Vol. 6, No. 8, 2001, pp. 427-436. doi:10.1016/S1359-6446(01)01757-3
- L. M. Kaminskas, V. M. McLeod, M. Victoria, B. D. Kelly. P. Karellas and C. J. H. Porter, “PEGylation of Polylysine Dendrimers Improves Absorption and Lymphatic Targeting Following SC Administration in Rats,” Journal of Controlled Release, Vol. 140, No. 2, 2009, pp. 108-116. doi:10.1016/j.jconrel.2009.08.005
- L. M. Kaminskas, B. J. Boyd, P. Karellas, S. A. Henderson, M. P. Giannis, G. Y. Krippner and C. H. J Porter, “Impact of Surface Derivatization of Poly-L-Lysine Dendrimers with Anionic Arylsulfonate or Succinate Groups on Intravenous Pharmacokinetics and Disposition,” Molecular Pharmaceutics, Vol. 4, No. 6, 2007, pp. 949-961. doi:10.1021/mp070047s
- B. J. Boyd, L. M. Kaminskas, P. Karellas, G. Y. Krippner, R. Lessene and C. H. J Porter, “Cationic Poly-L-Lysine Dendrimers: Pharmacokinetics, Biodistribution, and Evidence for Metabolism and Bioresorption after Intravenous Administration to Rats,” Molecular Pharmaceutics, Vol. 3, No. 5, 2006, pp. 614-627. doi:10.1021/mp060032e
- D. A Tomalia, H. Baker, J. Dewald, M. Hall, G. Kallos, S. Martin, J. Roeck, J. Ryder and P. Smith, “A New Class of Polymers: Starburst-Dendritic Macromolecules,” Polymer Journal, Vol. 17, No. 1, 1985, pp. 117-132. doi:10.1295/polymj.17.117
- K. Sadler and J. P. Tam, “Peptide Dendrimers: Applications and Synthesis,” Reviews in Molecular Biotechnology, Vol. 90, No. 3, 2002, pp. 195-229. doi:10.1016/S1389-0352(01)00061-7
- M. Monso, W. Kowalczyk, D. Andreu and B. G. de la Torre, “Reverse Thioether Ligation Route to Multimeric Peptide Antigens,” Organic & Biomolecular Chemistry, Vol. 10, No. 15, 2012, pp. 3116-3121. doi:10.1039/c2ob06819b
- F. A. Robey, R. L. Fields and W. Lindner, “Synthesis of Chloroacetyl and Bromoacetyl Modified Peptides for the Preparation of Synthetic Peptide Polymers, Conjugated Peptides, and Cyclic Peptides,” US Patents No. 5066716, 1991.
- A. Isidro-Llobet, M. Alvarez and F. Albericio, “Amino Acid-Protecting Groups,” Chemical Reviews, Vol. 109, No. 6, 2009, pp. 2455-2504. doi:10.1021/cr800323s
- K. Y. Win and S. S. Feng, “Effects of Particle Size and Surface Coating on Cellular Uptake of Polymeric Nanoparticles for Oral Delivery of Anticancer Drugs,” Biomaterials, Vol. 26, No. 15, 2005, pp. 2713-2722. doi:10.1016/j.biomaterials.2004.07.050
NOTES
*Corresponding author.