Paper Menu >>
Journal Menu >>
 J. Biomedical Science and Engineering, 2009, 2, 273-279 doi: 10.4236/jbise.2009.24041 Published Online August 2009 (http://www.SciRP.org/journal/jbise/ JBiSE ). Published Online August 2009 in SciRes. http:// www.scirp.org/journal/jbise Determination of inter- and intra-subtype/species varia- tions in polymerase acidic protein from influenza A virus using amino-acid pair predictability Shaomin Yan1, Guang Wu2* 1National Engineering Research Center for Non-food Biorefinery, Guangxi Academy of Sciences, 98 Daling Road, Nanning, Guangxi, 530007, China; 2Computational Mutation Project, DreamSciTech Consulting, 301, Building 12, Nanyou A-zone, Jiannan Road, Shenzhen, Guangdong Province 518054, China. Email: hongguanglishibahao@yahoo.com Received 23 March 2009; revised 15 April 2009; accepted 28 April 2009. ABSTRACT The polymerase acidic protein is an important family of proteins from influenza A virus, which is classified as many different subtypes or spe- cies. Thus, an important question is if these classifications are numerically distinguishable with respect to the polymerase acidic protein. The amino-acid pair predictability was used to transfer 2432 polymerase acidic proteins into 2432 scalar data. The one-way ANOVA found these polymerase acidic proteins distinguish- able in terms of subtypes and species. However, the large residuals in ANOVA suggested a pos- sible large intra-subtype/species variation. Therefore, the inter- and intra-subtype/species variations were studied using the model II ANOVA. The results showed that the in- tra-subtype/species variations accounted most of variation, which was 100% in total for both inter- and intra- subtype/species variations. Our analysis threw lights on the issue of how to de- termine a wide variety of patterns of antigenic variation across space and time, and within and between subtypes as well as hosts. Keywords: Amino-Acid Pair; Influenza A Virus; ln- ter- and Intra-; Model II ANOVA; Polymerase Acidic Protein; Species; Subtype; Variation 1. INTRODUCTION The unpredictable mutations in proteins from influenza A virus threaten the humans with possible pandemics or epidemics, therefore it is considered important to accu- rately, precisely and reliably predict the mutations. In this way, the new vaccines, which would be more effec- tive against the influenza A virus, could be manufactured [1,2,3,4,5,6]. Currently, the manufactured vaccines are designed to target the influenza A virus according to their subtypes, for example, the focus in recent year would be the H5N1 subtype of influenza A virus [7,8,9,10,11,12,13,14], and anti-flu drugs are designed to target neuraminidases and M2 protein [15,16]. It would be understandable that proteins should be different from one subtype to another. Otherwise, there would be no classification of subtype. Moreover, the proteins under the same subtype should be different one another, otherwise a single subtype would contain only a single protein. The same holds for pro- teins classified according to species, where the sample was obtained. Here, an important question is if these classifications are numerically distinguishable, say, if a protein is dif- ferent from species to species and from subtype to sub- type in number. This is the base for prediction of muta- tion using mathematical modeling. However this work has yet to be done, because the difference between proteins is different in terms of let- ters, which represent the amino acids in proteins. It is difficult to use any statistical method to determine these differences cross a protein family. For this aim, it needs to transfer a protein into a datum that should be different from protein to protein. Then it would be possible to conduct an ANOVA statistics to answer the question above. Actually there are quite a few methods, which can transfer a protein sequence into a series of numerical codes or numerical sequence for predicting its various attributes (see, e.g., [17,18,19,20,21,22,23,24,25]). Since 1999, we have developed three approaches to transfer each amino acid in a protein as well as a whole protein (for reviews, see [26,27,28]) into either a single datum or numerical sequence, which resulted in many studies on proteins. Afterward, another question would be the inter- and intra-subtype/species variations. This issue is important because the vaccines and anti-flu drugs manufactured based on subtype would be more efficient and effective if the difference within subtype/species would be smaller  274 S. M. Yan et al. / J. Biomedical Science and Engineering 2 (2009) 273-279 SciRes Copyright © 2009 JBiSE than that between subtypes/species. Influenza viruses replicate and transcribe their seg- mented negative-sense single-stranded RNA genome in the nucleus of the infected host cell. All RNA synthesiz- ing activities associated with influenza virus are per- formed by the virally encoded RNA-dependent RNA polymerase that consists of three subunits, polymerase acidic protein (PA), polymerase basic proteins 1 and 2 [29]. The PA subunit is involved for the conversion of RNA polymerase from transcriptase to replicase [30] and contains the endonuclease active site. A recent study strongly implicates the viral RNA polymerase complex as a major determinant of the pathogenicity of the 1918 pandemic virus [31]. Many studies have indicated that sequence-based pre- diction approaches, such as protein subcellular location prediction [32,33,34], protein quaternary attribute pre- diction [25,35], identification of membrane proteins and their types [36,37], identification of enzymes and their functional classes [38], identification of GPCR and their types [24,39,40], identification of proteases and their types [41,42], protein cleavage site prediction [43,44,45], signal peptide prediction [46,47], and protein 3D struc- ture prediction based on sequence alignment [48], can timely provide very useful information and insights for both basic research and drug design. The present study was attempted to use the model II ANOVA to investigate the inter- and intra- sub- type/species variations in polymerase acidic protein from influenza A virus in hope to shed lights on helping find effective drugs against influenza A virus. 2. MATERIALS AND METHODS 2.1. Data A total of 5165 full-length PA sequences of influenza A virus sampled from 1918 to 2008 was obtained from the influenza virus resources [49]. After excluded identical sequences, 2432 PA proteins were used in this study. 2.2. Transferring Symbolized PA Proteins into Scalar Data Among these methods developed by us, the amino-acid pair predictability is the simplest, which was thus used in this study. According to the permutation, the adjacent amino-acid pairs in a protein can be classified as predict- able and unpredictable, which provided a measure to dis- tinguish protein one another and was used in many our previous studies (for example 2008, [28,50,51,52,53,54]). For example, there was an avian influenza virus (strain A/quail/Hong Kong/1721-20/99(H6N1)) and its PA was composed of 716 amino acids (accession number CAC84865). The first and second amino acids could be counted as an amino-acid pair, the second and third as another amino-acid pairs, the third and fourth, until the 715th and 716th, thus there were 715 amino-acid pairs. Then, how many amino-acid pairs can be explained by the permutation or random mechanism in this PA? This can be determined using the percentage of predictable and unpredictable amino-acid pairs. There were 37 aspartic acids “D” and 76 glutamic ac- ids “E” in CAC84865 PA. If the permutation could ex- plain the appearance of amino-acid pair DE, it could appear four times in this PA (37/716×76/715×715=3.927). Actually there were 4 DEs in this PA. Thus, the appearance of DE could be explained by permutation or predicted by random mechanism. By clear contrast, there were 50 iso- leucines “I” in the PA. If the permutation could explain the appearance of IE, it could appear five times (50/716×76/715×715=5.307). However, it appeared 12 times in realty, which could not be explained by permuta- tion or randomly unpredictable. In this way, all amino-acid pairs in this PA could be classified as predictable and un- predictable. For this particular PA, its predictable and un- predictable portions were 25.45% and 74.55%. Taking another PA (accession number CAC84866) as example, this PA had only one amino acid different from CAC84865 PA at position 437. However, its predictable and unpredictable portions were 25.59% and 74.41%. Thus, the amino-acid pair predictability distinguished the difference between different PA proteins as a very sensitive measure. 2.3. Difference among Subtypes/Species Influenza A viruses are classified by the serological sub- types of the primary viral surface proteins. Currently, there are 16 haemagglutinin subtypes from H1 to H16 and 9 neuraminidase subtypes from N1 to N9 [55]. Also, influ- enza A viruses can be classified according to their host. After computation of 2432 PA proteins, the predict- able portions of PA proteins were grouped according to their classifications of subtypes and species. As there were more than two subtypes and two species, and the number of PA proteins was highly different from subtype to subtype, and from species to species, the one-way ANOVA followed by the Holm-Sidak’s compari- son test was used to compare the difference among and between subtypes/species using the SigmaStat software [56]. P < 0.05 was considered statistically significant. 2.4. Inter- and Intra-Subtype Variation The single classification model II ANOVA with unequal sample sizes [57] was used to determine the inter- and intra-subtype/species variations. 3. RESULTS AND DISCUSSION The one-way ANOVA showed that there were statisti- cally significant difference among HA subtypes (Figure 1), NA subtypes (Figure 2) and species (Figure 3). Even the statistical difference was found between subtypes and between species. The detailed results were listed in Supplementary results. 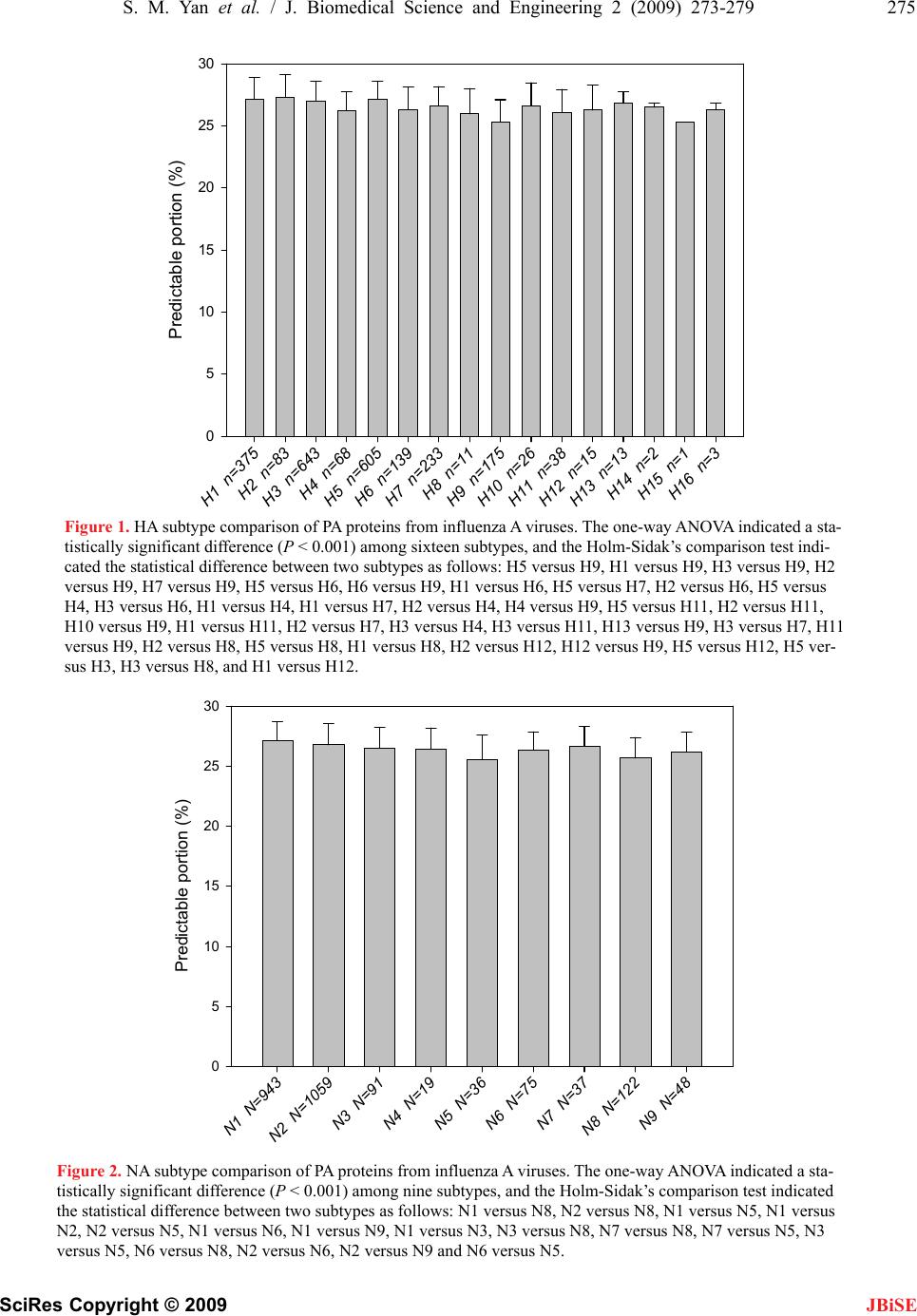 S. M. Yan et al. / J. Biomedical Science and Engineering 2 (2009) 273-279 275 SciRes Copyright © 2009 H1 n=375 H2 n=83 H3 n=643 H4 n=68 H5 n=605 H6 n=139 H7 n=233 H8 n=11 H9 n=175 H10 n=26 H11 n=38 H12 n=15 H13 n=13 H14 n=2 H15 n=1 H16 n=3 Predictable portion (%) 0 5 10 15 20 25 30 Figure 1. HA subtype comparison of PA proteins from influenza A viruses. The one-way ANOVA indicated a sta- tistically significant difference (P < 0.001) among sixteen subtypes, and the Holm-Sidak’s comparison test indi- cated the statistical difference between two subtypes as follows: H5 versus H9, H1 versus H9, H3 versus H9, H2 versus H9, H7 versus H9, H5 versus H6, H6 versus H9, H1 versus H6, H5 versus H7, H2 versus H6, H5 versus H4, H3 versus H6, H1 versus H4, H1 versus H7, H2 versus H4, H4 versus H9, H5 versus H11, H2 versus H11, H10 versus H9, H1 versus H11, H2 versus H7, H3 versus H4, H3 versus H11, H13 versus H9, H3 versus H7, H11 versus H9, H2 versus H8, H5 versus H8, H1 versus H8, H2 versus H12, H12 versus H9, H5 versus H12, H5 ver- sus H3, H3 versus H8, and H1 versus H12. N1 N=943 N2 N=1059 N3 N=91 N4 N=19 N5 N=36 N6 N=75 N7 N=37 N8 N=122 N9 N=48 Predictable portion (%) 0 5 10 15 20 25 30 Figure 2. NA subtype comparison of PA proteins from influenza A viruses. The one-way ANOVA indicated a sta- tistically significant difference (P < 0.001) among nine subtypes, and the Holm-Sidak’s comparison test indicated the statistical difference between two subtypes as follows: N1 versus N8, N2 versus N8, N1 versus N5, N1 versus N2, N2 versus N5, N1 versus N6, N1 versus N9, N1 versus N3, N3 versus N8, N7 versus N8, N7 versus N5, N3 versus N5, N6 versus N8, N2 versus N6, N2 versus N9 and N6 versus N5. JBiSE 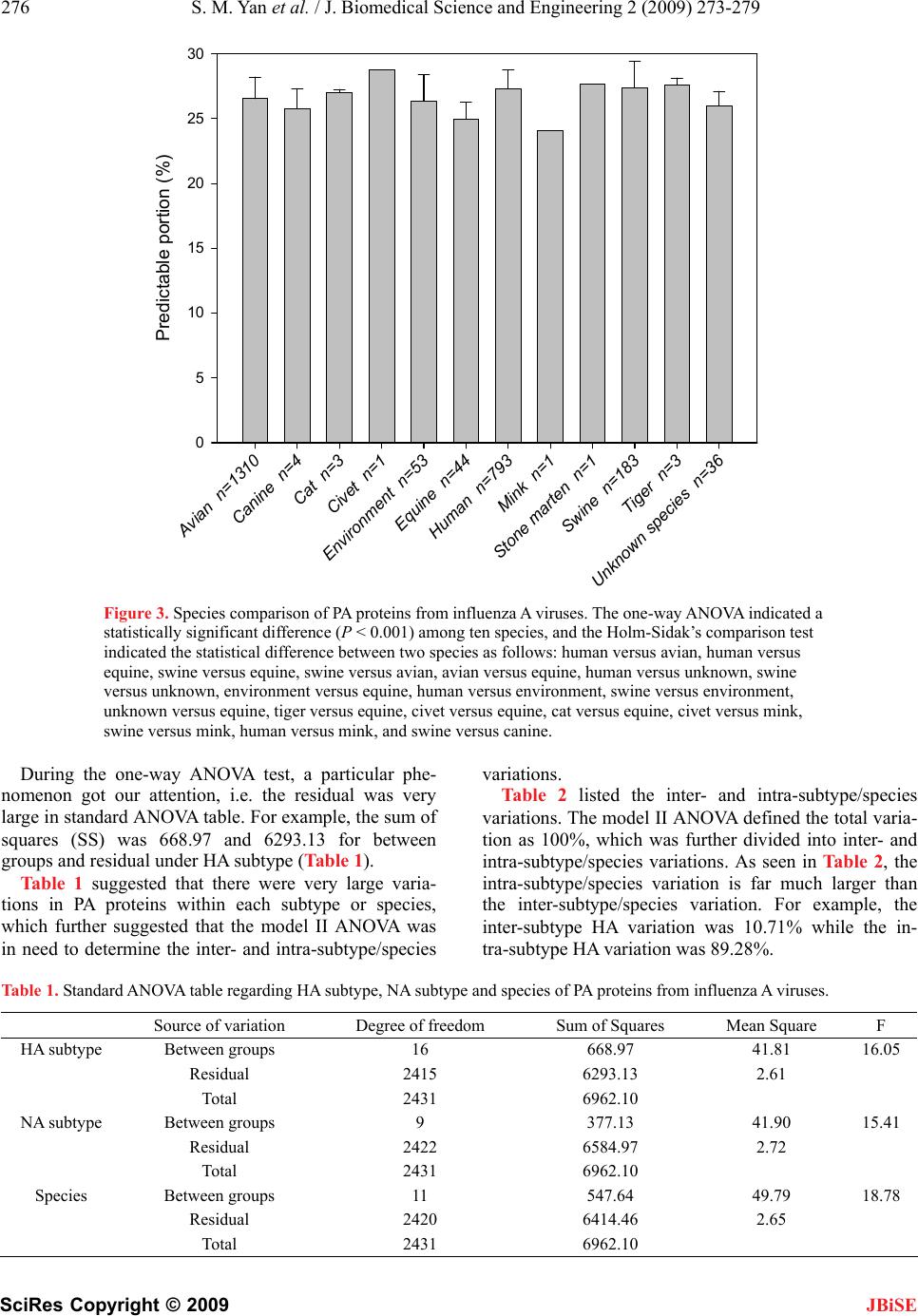 276 S. M. Yan et al. / J. Biomedical Science and Engineering 2 (2009) 273-279 SciRes Copyright © 2009 Avian n=1310 Canine n=4 Cat n=3 Civet n=1 Environment n=53 Equine n=44 Human n=793 Mink n=1 Stone marten n=1 Swine n=183 Tiger n=3 Unknown species n=36 Predictable portion (%) 0 5 10 15 20 25 30 Figure 3. Species comparison of PA proteins from influenza A viruses. The one-way ANOVA indicated a statistically significant difference (P < 0.001) among ten species, and the Holm-Sidak’s comparison test indicated the statistical difference between two species as follows: human versus avian, human versus equine, swine versus equine, swine versus avian, avian versus equine, human versus unknown, swine versus unknown, environment versus equine, human versus environment, swine versus environment, unknown versus equine, tiger versus equine, civet versus equine, cat versus equine, civet versus mink, swine versus mink, human versus mink, and swine versus canine. During the one-way ANOVA test, a particular phe- nomenon got our attention, i.e. the residual was very large in standard ANOVA table. For example, the sum of squares (SS) was 668.97 and 6293.13 for between groups and residual under HA subtype (Table 1). Table 1 suggested that there were very large varia- tions in PA proteins within each subtype or species, which further suggested that the model II ANOVA was in need to determine the inter- and intra-subtype/species variations. Table 2 listed the inter- and intra-subtype/species variations. The model II ANOVA defined the total varia- tion as 100%, which was further divided into inter- and intra-subtype/species variations. As seen in Table 2, the intra-subtype/species variation is far much larger than the inter-subtype/species variation. For example, the inter-subtype HA variation was 10.71% while the in- tra-subtype HA variation was 89.28%. Table 1. Standard ANOVA table regarding HA subtype, NA subtype and species of PA proteins from influenza A viruses. Source of variation Degree of freedom Sum of Squares Mean Square F HA subtype Between groups 16 668.97 41.81 16.05 Residual 2415 6293.13 2.61 Total 2431 6962.10 NA subtype Between groups 9 377.13 41.90 15.41 Residual 2422 6584.97 2.72 Total 2431 6962.10 Species Between groups 11 547.64 49.79 18.78 Residual 2420 6414.46 2.65 Total 2431 6962.10 JBiSE  S. M. Yan et al. / J. Biomedical Science and Engineering 2 (2009) 273-279 277 SciRes Copyright © 2009 JBiSE Table 2. Inter- and intra-subtype/species variations of PA pro- teins from influenza A viruses. Classification Inter-subtype/ species variation Intra-subtype/ species variation HA subtype 10.71% 89.28% NA subtype 7.54% 92.46% Species 11.88% 88.12% At this point, one might wonder why the statistical difference was found among subtypes and species while there were so large variations within subtype and species. These results in fact were very reasonable. In plain words, there would be statistical difference between males and females in performing a sport, for example, however the difference between male sportsman and ordinary male would be also huge, thus it could be pos- sible this variation would be larger than that between males and females. In fact, the single classification model II ANOVA has many important applications although this method is less familiar with most researchers [58,59]. For example, it is better to know the inter- and intra-patient variations be- fore planning clinical experiments, say, if an experimen- tal design should include two parallel groups (inter- pa- tients) or a two-part crossover design (intra-patients). In the context of this study, generally, a small intra- subtype/species variation suggested a cost-effective way in collecting of samples, it said, not many samples for a particular subtype/species were in need, by clear contrast, many samples were in need regarding a particular sub- type/species if there was a large intra- subtype/species variation. Actually, the inter- and intra-subtype/species varia- tions were the mutations occurred in the same subtype or species, and occurred cross subtypes or species. There was a wide variety of patterns of antigenic variation across space and time, and within and between subtypes as well as hosts and we did not yet understand the de- terminants of these different patterns [60]. Our analysis could shed lights on this issue. The far much large intra-subtype/species variation found in this study suggested: 1) much more PA proteins belonging to the same subtype/species were needed in order to better understand the mutation pattern in the same subtype/species, and 2) the current classification was based on the surface proteins from influenza A virus, while under the same subtype, the PA mutations were quite large. On the other hand, the statistically significant differ- ence between subtypes suggested the current classifica- tion valid even for the PA, which was an internal protein. The most important requirement for producing vaccines against viruses displaying antigenic diversity is a method of measuring antigenic distances between strains and developing an understanding of how these distances re- late to cross-protection [61]. The current results sup- ported the idea to develop vaccines and anti-flu drugs that generate effective heterosubtypic immunity based on immune recognition of influenza A virus antigens conserved across all viral strains [62,63]. 4. ACKNOWLEDGEMENTS This study was partly supported by International Science & Technology Cooperation Projects (2008DFA30710), Guangxi Science Foundation (No. 0630003A2 and 0991080), and Guangxi Academy of Sciences (project No. 09YJ17 SW07). REFERENCES [1] I. Capuaa and D. J. Alexander, (2008) Avian influenza vaccines and vaccination in birds, Vaccine, 26(4), D70-D73. [2] N. Hehme, T. Colegate, B Palache, and L. Hessel, (2008) Influenza vaccine supply: Building long-term sustain- ability, Vaccine, 26(4), D23-D26. [3] M. Hoelscher, S. Gangappa, W. Zhong, L. Jayashankar, and S. Sambhara, (2008) Vaccines against epidemic and pandemic influenza, Expert Opin. Drug Deliv., 5, 1139 -1157. [4] B. Palache, (2008) New vaccine approaches for seasonal and pandemic influenza, Vaccine, 26, 6232-6236. [5] J. C. Tilburt, P. S. Mueller, A. L. Ottenberg, G. A. Poland, and B. A. Kioenig, (2008) Facing the challenges of in- fluenza in healthcare settings: The ethical rationale for mandatory seasonal influenza vaccination and its impli- cations for future pandemics, Vaccine, 26(4), D27-D30. [6] M. Peyre, G. Fusheng, S. Desvaux, and F. Roger, (2009) Avian influenza vaccines: A practical review in relation to their application in the field with a focus on the Asian experience, Epidemiol. Infect., 137, 1-21. [7] A. W. Hampson, (2008) Vaccines for pandemic influenza: The history of our current vaccines, their limitations and the requirements to deal with a pandemic threat, Ann. Acad. Med. Singapore, 37, 510-517. [8] N. Skeik and F. I. Jabr, (2008) Influenza viruses and the evolution of avian influenza virus H5N1, Int. J. Infect. Dis., 12, 233-238. [9] D. Q. Wei, Q. S. Du, H. Sun, and K. C. Chou, (2006) Insights from modeling the 3D structure of H5N1 influ- enza virus neuraminidase and its binding interactions with ligands, Biochem. Biophys. Res. Commun., 344, 1048-1055. [10] S. Q. Wang, Q. S. Du, and K. C. Chou, (2007) Study of drug resistance of chicken influenza A virus (H5N1) from homology-modeled 3D structures of neuramini- dases, Biochem. Biophys. Res. Commun., 354, 634-640. [11] Q. S. Du, S. Q. Wang, and K. C. Chou, (2007) Analogue inhibitors by modifying oseltamivir based on the crystal neuraminidase structure for treating drug-resistant H5N1 virus, Biochem. Biophys. Res. Commun., 362, 525-531. [12] X. L. Guo, L. Li, D. Q. Wei, Y. S. Zhu, and K. C. Chou,  278 S. M. Yan et al. / J. Biomedical Science and Engineering 2 (2009) 273-279 SciRes Copyright © 2009 JBiSE (2008) Cleavage mechanism of the H5N1 hemagglutinin by trypsin and furin. Amino Acids, 35, 375-382. [13] R. B. Huang, Q. S. Du, C. H. Wang, and K. C. Chou, (2008) An in-depth analysis of the biological functional studies based on the NMR M2 channel structure of in- fluenza A virus, Biochem. Biophys. Res. Comm., 377, 1243-1247. [14] Q. S. Du, R. B. Huang, C. H. Wang, X. M. Li, and K. C. Chou, (2009) Energetic analysis of the two controversial drug binding sites of the M2 proton channel in influenza A virus, J. Theoret. Biol., doi:10.1016/j.jtbi.2009.1003. 1003. [15] J. R. Schnell, and J. J. Chou, (2008) Structure and mechanism of the M2 proton channel of influenza A vi- rus, Nature, 451, 591-595. [16] R. M. Pielak, J. R. Jason, R. Schnell, and J. J. Chou, (2009) Mechanism of drug inhibition and drug resistance of influenza A M2 channel, Proc. Natl. Acad. Sci. USA, www. pnas.org cgi doi 10.1073 pnas.0902548106. [17] K. C. Chou, (2001) Prediction of protein cellular attrib- utes using pseudo amino acid composition, PROTEINS: Structure, Function, and Genetics (Erratum: ibid., 2001, Vol.44, 60), 43, 246-255. [18] K. C. Chou, (2005) Using amphiphilic pseudo amino acid composition to predict enzyme subfamily classes, Bioinformatics, 21, 10-19. [19] X. Xiao, S. H. Shao, Z. D. Huang, and K. C. Chou, (2006) Using pseudo amino acid composition to predict protein structural classes: Approached with complexity measure factor, J. Comput. Chem., 27, 478-482. [20] X. Xiao, S. Shao, Y. Ding, Z. Huang, Y. Huang, and K. C. Chou, (2005) Using complexity measure factor to predict protein subcellular location, Amino Acids, 28, 57-61. [21] X. Xiao and K. C. Chou, (2007) Digital coding of amino acids based on hydrophobic index, Protein Pept. Lett., 14, 871-875. [22] X. Xiao, W. Z. Lin, and K. C. Chou, (2008) Using grey dynamic modeling and pseudo amino acid composition to predict protein structural classes, J. Comput. Chem., 29, 2018-2024. [23] X. Xiao, P. Wang, and K. C. Chou, (2008) Predicting protein structural classes with pseudo amino acid com- position: An approach using geometric moments of cel- lular automaton image, J. Theoret. Biol., 254, 691-696. [24] X. Xiao, P. Wang, and K. C. Chou, (2008) GPCR-CA: A cellular automaton image approach for predicting G- pro- tein-coupled receptor functional classes, J. Comput. Chem., DOI 10.1002/jcc.21163. [25] X. Xiao, P. Wang, and K. C. Chou, (2009) Predicting protein quaternary structural attribute by hybridizing functional domain composition and pseudo amino acid composition, J. Appl. Crystallogr., 42, 169-173. [26] G. Wu and S. Yan, (2002) Randomness in the primary structure of protein: Methods and implications, Mol. Biol. Today, 3, 55-69. [27] G. Wu and S. Yan, (2006) Mutation trend of hemaggluti- nin of influenza A virus: a review from computational mutation viewpoint, Acta Pharmacol. Sin., 27, 513-526. [28] G. Wu and S. Yan, (2008) Lecture notes on computational mutation, Nova Science Publishers, New York. [29] O. G. Engelhardt and E. Fodor, (2006) Functional asso- ciation between viral and cellular transcription during in- fluenza virus infection, Rev. Med. Virol., 16, 329-345. [30] A. Honda and A. Ishihama, (1997) The molecular anat- omy of influenza virus RNA polymerase, Biol. Chem., 378, 483-488. [31] T. Watanabe, S. Watanabe, K. Shinya, J. H. Kim, M. Hatta, and Y. Kawaoka, (2009) Viral RNA polymerase complex promotes optimal growth of 1918 virus in the lower respiratory tract of ferrets, Proc. Natl. Acad. Sci. USA, 106, 588-592. [32] K. C. Chou and H. B. Shen, (2008) Cell-PLoc: A package of web-servers for predicting subcellular localization of proteins in various organisms, Nature Prot., 3, 153-162. [33] K. C. Chou and H. B. Shen, (2007) Review: Recent pro- gresses in protein subcellular location prediction, Analyt. Biochem., 370, 1-16. [34] K. C. Chou and H. B. Shen, (2007) Euk-mPLoc: A fusion classifier for large-scale eukaryotic protein subcellular location prediction by incorporating multiple sites, J. Proteome Res., 6, 1728-1734. [35] H. B. Shen and K. C. Chou, (2009) QuatIdent: A web server for identifying protein quaternary structural attrib- ute by fusing functional domain and sequential evolution information, J. Proteome Res., 8, 1577-1584. [36] K. C. Chou and H. B. Shen, (2007) MemType-2L: A Web server for predicting membrane proteins and their types by incorporating evolution information through Pse- PSSM, Biochem. Biophys. Res. Commun., 360, 339-345. [37] T. Wang, J. Yang, H. B. Shen, and K. C. Chou, (2008) Predicting membrane protein types by the LLDA algo- rithm, Protein Pept. Lett., 15, 915-921. [38] H. B. Shen and K. C. Chou. (2007) EzyPred: A top-down approach for predicting enzyme functional classes and subclasses, Biochem. Biophys. Res. Commun., 364, 53- 59. [39] K. C. Chou and D. W. Elrod, (2002) Bioinformatical analysis of G-protein-coupled receptors, J. Proteome Res., 1, 429-433. [40] K. C. Chou, (2005) Prediction of G-protein-coupled re- ceptor classes, J. Proteome Res., 4, 1413-1418. [41] K. C. Chou and H. B. Shen, (2008) ProtIdent: A web server for identifying proteases and their types by fusing functional domain and sequential evolution information, Biochem. Biophys, Res. Comm., 376, 321-325. [42] H. B. Shen and K. C. Chou. (2009) Identification of pro- teases and their types, Analyt. Biochem., 385, 153-160. [43] K. C. Chou, (1993) A vectorized sequence-coupling model for predicting HIV protease cleavage sites in pro- teins, J. Biol. Chem., 268, 16938-16948. [44] K. C. Chou, (1996) Review: Prediction of HIV protease cleavage sites in proteins, Analyt. Biochem., 233, 1-14. [45] H. B. Shen and K. C. Chou, (2008) HIVcleave: a web- server for predicting HIV protease cleavage sites in pro- teins, Analyt. Biochem., 375, 388-390. [46] K. C. Chou and H. B. Shen, (2007) Signal-CF: A subsite- coupled and window-fusing approach for predicting sig- nal peptides, Biochem. Biophys. Res. Commun., 357, 633-640.  S. M. Yan et al. / J. Biomedical Science and Engineering 2 (2009) 273-279 279 SciRes Copyright © 2009 JBiSE [47] H. B. Shen and K. C. Chou, (2007) Signal-3L: A 3-layer approach for predicting signal peptide, Biochem. Bio- phys. Res. Commun., 363, 297-303. [48] K. C. Chou, (2004) Review: Structure bioinformatics and its impact to biomedical science, Curr. Med. Chem., 11, 2105-2134. [49] R. Apweiler, A. Bairoch and C. H. Wu. (2005) Protein sequence databases, Curr. Opin. Chem. Biol., 8, 76-80. [50] G. Wu and S. Yan, (2008) Prediction of mutations engi- neered by randomness in H5N1 neuraminidases from in- fluenza A virus, Amino Acids, 34, 81-90. [51] G. Wu and S. Yan, (2008) Prediction of mutations initi- ated by internal power in H3N2 hemagglutinins of influ- enza A virus from North America, Int. J. Pept. Res. Ther., 14, 41-51. [52] G. Wu and S. Yan, (2008) Prediction of mutation in H3N2 hemagglutinins of influenza A virus from North America based on different datasets, Protein Pept. Lett., 15, 144-152. [53] G. Wu and S. Yan, (2008) Three sampling strategies to predict mutations in H5N1 hemagglutinins from influ- enza A virus, Protein Pept. Lett., 15, 731-738. [54] G. Wu and S. Yan. (2008) Prediction of mutations engi- neered by randomness in H5N1 hemagglutinins of influ- enza A virus, Amino Acids, 35, 365-373. [55] E. Spackman, (2008) A brief introduction to the avian influenza virus, Methods Mol. Biol., 436, 1-6. [56] SPSS Inc. (1992-2003) SigmaStat for windows version 3.00. [57] R. R. Sokal and F. J. Rohlf, (1995) Biometry: The princi- ples and practices of statistics in biological research, 2nd ed, W. H. Freeman, New York, 203-218. [58] G, Wu, M. Baraldo, and M. Furlanut, (1999) Inter-patient and intra-patient variations in the baseline tapping test in patients with Parkinson’s disease, Acta Neurol. Belg., 99, 182-184. [59] M. Furlanut, G. Wu, and E. Perucca, (2001) Variability in the metabolism of levodopa and clinical implications, In: Interindividual Variability in Drug Metabolism in Man. eds. by Pacifici GM, Pelkonen O, Tayler & Francis, London and New York, Chapter 7, 181-227. [60] M. Lipsitch and J. J. O’Hagan, (2007) Patterns of anti- genic diversity and the mechanisms that maintain them, J. R. Soc. Interface, 4, 787-802. [61] J. A. Mumford, (2007) Vaccines and viral antigenic di- versity, Rev. Sci. Tech., 26, 69-90. [62] F. Carrat and A. Flahault, (2007) Influenza vaccine: The challenge of antigenic drift, Vaccine, 25, 6852-6862. [63] K. M. Grebe, J. W. Yewdell, and J. R. Bennink, (2008) Heterosubtypic immunity to influenza A virus, where do we stand? Microbes. Infect., 10, 1024-1029. |