 International Journal of Organic Chemistry, 2011, 1, 26-32 doi:10.4236/ijoc.2011.12005 Published Online June 2011 (http://www.SciRP.org/journal/ijoc) Copyright © 2011 SciRes. IJOC A Simple and Highly Efficient Enantioselective Synthesis of (S)-Rivastigmine Veera R. Arava1*, Laxminarasimhulu Gorentla1, Pramod K. Dubey2 1R&D Laboratory, Suven Life Sciences Ltd., Hyderabad, India 2Department of Chemistry, J. N. T. University, Hyderabad, India E-mail: reddyvenis@rediffmail.com Received April 1, 2011; revised May 17, 2011; accepted May 28, 2011 Abstract A highly efficient and convenient procedure for the enantioselective synthesis of (S)-Rivastigmine, a cho- linergic agent for the treatment of mild to moderate dementia of the Alzheimer’s type and dementia due to Parkinson’s disease, is accomplished by the treatment of versatile, readily accessible (S)-(-)-2-methyl- 2-propanesulfinamide with 3-hydroxyacetophenone. This protocol provides high yield and excellent enan- tiomeric excess in short step synthesis. Keywords: Cholinergic Agent, Enantioselective, Highly Efficient, (S)-(-)-2-Methyl-2-Propane Sulfinamide, (S)-Rivastigmine 1. Introduction Alzheimer’s disease (AD) is the most common form of dementia, a severe human health threat with more than 30 million sufferers worldwide [1]. Rivastigmine, (S)-3- [1-(dimethylamino)ethyl] phenyl ethyl (methyl) car- bamate 1, is the first USFDA approved drug in the form of capsules and patches for the treatment of mild to moderate dementia of the Alzheimer’s type [2-5] and for mild to moderate dementia related to Parkinson’s disease [6]. In the year 2006, it has been used in more than 6 million patients worldwide. Rivastigmine works by in- hibiting both butyrylcholinesterase (BuChE) and acetyl- cholinesterase (AChE) with the same potency, unlike donepezil 2 (which selectively inhibits acetylcholines- terase), mimantine 3 and galanthamine 4. Its pharmacol- ogical effect is selectively on the central nervous system, preferentially on the monomeric G1 isoform of AChE (selectivity for different isoforms of BuChE have not yet been investigated). The metabolism of rivastigmine is by the enzyme cholinesterase and do not depend on the P450 system in the liver, which reduces the risk of inter- actions with other medications [7]. The other stigmine products are pyridostigmine 5, physostigmine 6 and neo- stigmine 7 (Figure 1). Rivastigmine has one chiral center with amine func- tionality. Therefore, attention needs to be paid to the synthesis of the correct enantiomer. (S)-enantiomer ex- hibits the desired cholinesterase inhibition, which re- quires the drug in enantiomerically pure form. The insertion of chiral amine functionality through N-sulfinylimines is a major breakthrough endeavor due to the exceptional behavior of the chiral sulfinyl group in N-sulfinylimines, as an activator, chiral controller and useful protective group and finally recyclability [8] makes the sulfinamides extremely versatile chiral re- agents (Figure 2) [9]. 2. Results and Discussion Our research group has been interested in utilization of these sulfinamides in industrial perspective for the de- velopment of facile chemical processes for active phar- maceutical ingredients (API’s) [10]. Although several methods for the synthesis of rivastigmine and phenyl- carbamate derivatives have been reported (Scheme 1), these methods suffer from limitations. Initial approaches have been developed via resolution of recemate using chiral acids [11-14] and transition metal catalysis [15,16]. Recent approaches are based on lipase catalyzed kinetic resolution [17-19], chemoenzymatic asymmetric synthe- sis [20] and asymmetric transfer hydrogenation [21]. In order to circumvent these difficulties and provide access to feasible industrial process, we have devised a highly efficient enantioselective synthetic route based on the report that enantiopure tert-butane-sulfinamides take 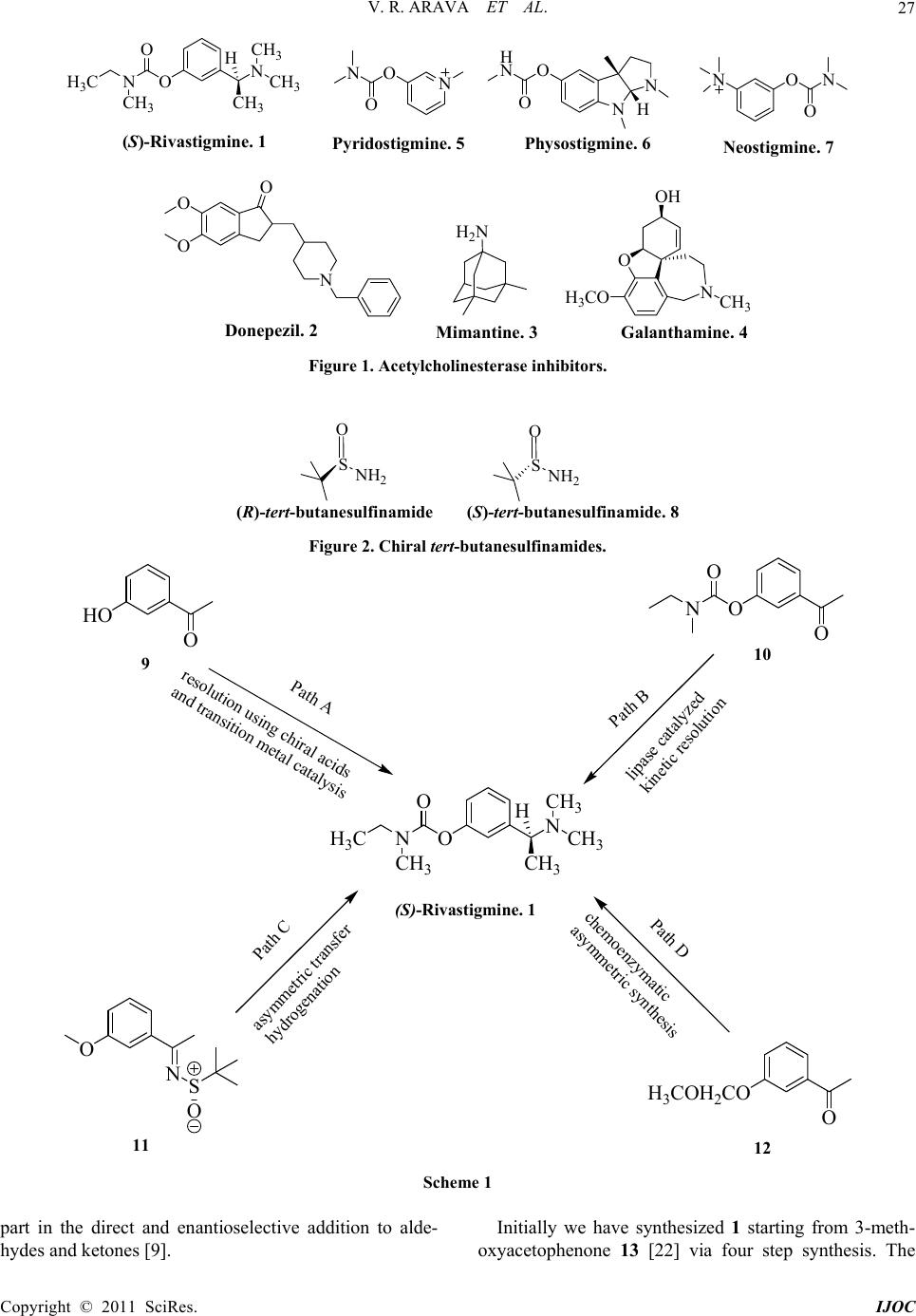 V. R. ARAVA ET AL.27 ON NH 3 C CH 3 CH 3 H CH 3 CH 3 O (S)-Rivastigmine. 1 N ON O Pyrid osti g mine. 5 N N O H N OH Physostigmine . 6 O N O N Neostigmine. 7 O O O N Donepezil. 2 H 2 N Mimantine. 3Galanthamine. 4 OH O NCH 3 H 3 CO Figure 1. Acetylcholinesterase inhibitors. SNH 2 O SNH 2 O (R)-tert-butanesulfinamide (S)-tert-butanesulfinamide. 8 Figure 2. Chiral tert-butanesulfinamides. ON NH 3 C CH 3 CH 3 H CH 3 CH 3 O (S)-Rivastigmine. 1 HO O 9 O NS O 11 H 3 COH 2 CO O 12 O O N O 10 Path A Path B Path C Path D resolution using chiral acids and transition metal catalysis lipase catalyzed kinetic resolution asymmetric transfer hydrogenation chemoenzymatic asymmetric synthesis Scheme 1 part in the direct and enantioselective addition to alde- hydes and ketones [9]. Initially we have synthesized 1 starting from 3-meth- oxyacetophenone 13 [22] via four step synthesis. The Copyright © 2011 SciRes. IJOC 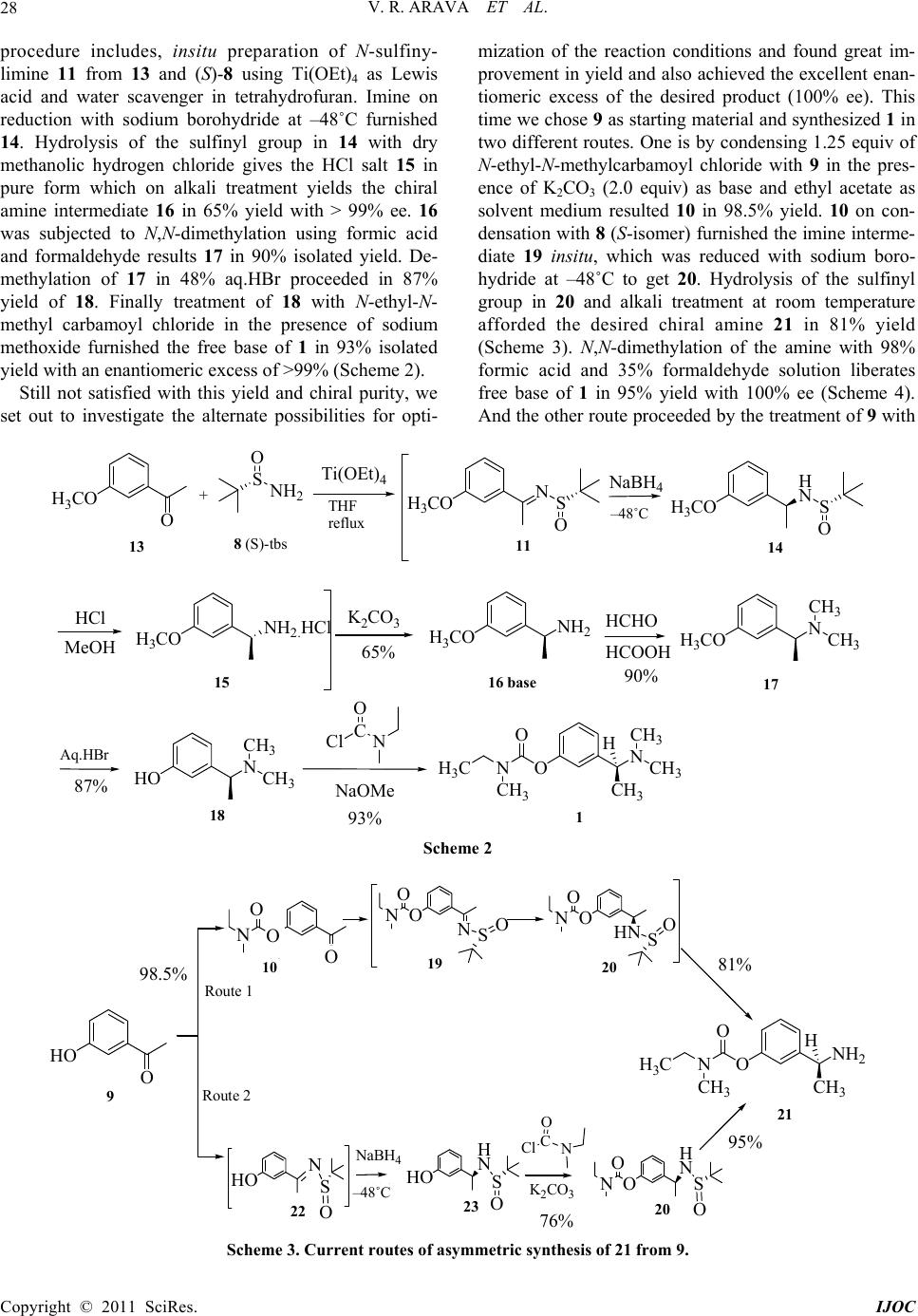 V. R. ARAVA ET AL. 28 procedure includes, insitu preparation of N-sulfiny- limine 11 from 13 and (S)-8 using Ti(OEt)4 as Lewis acid and water scavenger in tetrahydrofuran. Imine on reduction with sodium borohydride at –48˚C furnished 14. Hydrolysis of the sulfinyl group in 14 with dry methanolic hydrogen chloride gives the HCl salt 15 in pure form which on alkali treatment yields the chiral amine intermediate 16 in 65% yield with > 99% ee. 16 was subjected to N,N-dimethylation using formic acid and formaldehyde results 17 in 90% isolated yield. De- methylation of 17 in 48% aq.HBr proceeded in 87% yield of 18. Finally treatment of 18 with N-ethyl-N- methyl carbamoyl chloride in the presence of sodium methoxide furnished the free base of 1 in 93% isolated yield with an enantiomeric excess of >99% (Scheme 2). Still not satisfied with this yield and chiral purity, we set out to investigate the alternate possibilities for opti- mization of the reaction conditions and found great im- provement in yield and also achieved the excellent enan- tiomeric excess of the desired product (100% ee). This time we chose 9 as starting material and synthesized 1 in two different routes. One is by condensing 1.25 equiv of N-ethyl-N-methylcarbamoyl chloride with 9 in the pres- ence of K2CO3 (2.0 equiv) as base and ethyl acetate as solvent medium resulted 10 in 98.5% yield. 10 on con- densation with 8 (S-isomer) furnished the imine interme- diate 19 insitu, which was reduced with sodium boro- hydride at –48˚C to get 20. Hydrolysis of the sulfinyl group in 20 and alkali treatment at room temperature afforded the desired chiral amine 21 in 81% yield (Scheme 3). N,N-dimethylation of the amine with 98% formic acid and 35% formaldehyde solution liberates free base of 1 in 95% yield with 100% ee (Scheme 4). And the other route proceeded by the treatment of 9 with + S O NH 2 8 (S)-tbs Ti(OEt) 4 NaBH 4 -48°C H 3 CO O H 3 CO NS O H 3 CO H NS O H 3 CO NH 2. HCl 11 14 15 THF reflux 13 ON NH 3 C CH 3 CH 3 H CH 3 CH 3 O HCHO HCOOH C O Cl N NaOMe H 3 CO NH 2 H 3 CO N CH 3 CH 3 HO N CH 3 CH 3 1 17 18 16 base K 2 CO 3 HCl MeOH 48˚C Aq.HBr 65% 90% 87% 93% Scheme 2 C O Cl N HO O 9 ONH 2 NH 3 C CH 3 CH 3 H O 21 NaBH 4 -48°C HO N S O HO H N S O 22 23 K 2 CO 3 O H NS O O N 20 OO O NON O N SOOHN O N SO Route 1 Route 2 10 19 20 O 98.5% 81% 76% 95% Scheme 3. Current routes of asymmetric synthesis of 21 from 9. 48˚C 19 20 10 O O Copyright © 2011 SciRes. IJOC  V. R. ARAVA ET AL.29 ONH 2 NH 3 C CH 3 CH 3 H O 21 ON NH 3 C CH 3 CH 3 H CH 3 CH 3 O 1 HCHO HCOOH Scheme 4 8 (S-isomer) first, then reduction of the N-sulfinylimine 22 followed by amido-ester formation with N-ethyl-N- methyl carbamoyl chloride to afford 20 in 76% yield. Hydrolysis of sulfinyl group with dry methanolic hydro- gen chloride and alkali treatment gives 21 base with chiral purity > 94% in 95% yield (Scheme 3). Finally N,N-dimethylation of amine 21 with formic acid and 35% formaldehyde solution furnished 1 (free base) with 98% chiral purity in 95% yield (Scheme 4). The chiral purity is then improved to 100% by tartarate salt prepa- ration with L-(+)-tartaric acid. 3. Experimental Section Experiments are conducted under nitrogen atmosphere unless stated otherwise. All solvents and reagents are reagent grade pure and used without further purification. All melting points are determined on Polmon MP_96 melting point apparatus. 1H & 13C NMR spectra are re- corded using a Bruker 400 MHz spectrometer (400 & 100 MHz respectively) with TMS as internal standard. Mass spectra are recorded on a Perkin-Elmer mass spec- trometer operating at an ionization potential of 70 eV. IR spectra are recorded on Perkin Elmer spectrophotometer as KBr pellets or neat. Analytical TLC is conducted on E-Merck 60F254 aluminum-packed silicagel plates (0.2 mm). Developed plates are visualized under UV light or Iodine chamber. Chiral HPLC spectra are recorded on Waters alliance 2695 with 2487 U. V. detector. Typical procedure for the preparation of (S)-Rivasti- gmine (1) Route A. Synthesis of 3-acetylphenyl ethyl (methyl) car- bamate (10): a mixture of 9 (50.0 g, 0.367 mol), K2CO3 (101.5 g, 0.734 mol) and N-ethyl-N-methyl carbamoyl chloride (56.0 g, 0.46 mol) in ethyl acetate (500 ml) is refluxed for 4 hours. (Completion of reaction is moni- tored by TLC). The reaction mixture is cooled to room temperature and washed with water (3 × 250 mL), then dried over anhydrous Na2SO4, filtered and evaporated to get pale yellow oil of 10. Wt 80.0 g, 98.5% yield; HPLC Purity: 97%; IR (Neat, νmax, cm–1): 1724 (C = O), 1686 (C = O), 2974 (aliphatic CH); 1H-NMR (400MHz, CDCl3): δH 7.74 (d, 1H, J = 7.6 Hz, ArH), 7.65 (s, 1H, ArH), 7.40 (t, 1H, J = 7.9 Hz, ArH), 7.30 (d, 1H, J = 7.5 Hz, ArH), 3.42 (q, 1H, J = 7.0 Hz, rotamer1 CH2), 3.37 (q, 1H, J = 7.0 Hz, rotamer2 CH2), 3.04 (s, 1.5H, ro- tamer1 CH3), 2.95 (s, 1.5H, rotamer2 CH3), 2.55 (s, 3H, CH3), 1.21 (t, 1.5H, J = 7.0 Hz, rotamer1 CH3), 1.15 (t, 1.5H, J = 7.0 Hz, rotamer2 CH3); 13C NMR (100MHz, CDCl3): δC 197.08 (1C, C=O), 153.99 (d, J = 15.0 Hz, 1C, C=O), 151.64 (Caryl), 138.19 (Caryl), 129.26 (Caryl), 126.57 (Caryl), 124.93 (Caryl), 121.47 (Caryl), 43.98 (1C, CH2 of carbamoyl), 34.14 (rotamer1 Mecarbamoyl), 33.70 (rotamer2 Mecarbamoyl), 26.56 (1C, CH3), 13.10 (rotamer1 Mecarbamoyl), 12.29 (rotamer2 Mecarbamoyl); ESI-MS m/z (%): 222.0 (M+, 100), 223.1 (M+ + H, 20). Synthesis of (S)-Ethyl-methyl-carbamic acid 3-(1-aminoethyl) phenyl ester (21): compound 10 (80.0 g, 0.36 mol) is refluxed with (S)-tert-butanesul- finamide 8 (48.2 g, 0.39 mol), titanium tetra ethoxide (164.26 g, 0.72 mol) in tetrahydrofuran (800.0 ml) for 30 hours. Cool to ambient temperature and further to –48 to –52˚C. Add NaBH4 (20.5 g, 0.54 mol) in 5 portions. Stir for 3 hours to complete the reduction of imine intermedi- ate 19. Gradually warm to –5˚C, add methanol (80 ml) drop wise at –5˚C to 0˚C and stir for 30 minutes. Add ethyl acetate (400 ml) and water (500 ml). Stir for 30 minutes to warm to rt and filter off the salts. Separate the organic layer and aqueous layer is washed with ethyl acetate (200 ml). Combined organic layers are washed successively with water (2 × 250 ml) and brine solution (200 ml). The organic layer is dried over anhydrous Na2SO4 and evaporated under reduced pressure to get yellow oil of crude 20, which is hydrolyzed with 13% methanolic HCl (126 ml) and then alkali treatment with aq.NaOH at rt, extraction into ethyl acetate and concen- tration of the solvent afford pale yellow oil. Wt 65.0 g, yield 81%; [α]D 20 – 16.5˚ (c 1.0, CHCl3); Chiral HPLC Purity: 99.9%, Column: chiral cel OD-H 250 × 4.6 (SRC-583), mobile phase A: n-hexane:IPA:DEA (90:10: 0.1), Iso: 100%, concentration: 10 mg/10mL, diluent: ethanol, run time: 20 min, temperature: 25˚C, flow rate: 1 mL/min, UV: 225nm, retention time: (S)-isomer: 12.8 min and (R)-isomer: 11.66 min; HPLC Purity: 98.8%; IR (Neat, νmax, cm–1): 1716 (C=O), 2974 (aliphatic CH), 3365 (NH2); 1H-NMR (400MHz, CDCl3): δH 7.31 (t, 1H, J = 7.8 Hz, ArH), 7.27 (s, 1H, ArH), 7.16 (d, 1H, J = 7.7 Hz, ArH), 6.99 (d, 1H, J = 7.7 Hz, ArH), 4.12 (q, 1H, J = 3.4 Hz, CH), 3.46 (q, 1H, J = 7.2 Hz, rotamer1 CH2), Copyright © 2011 SciRes. IJOC  V. R. ARAVA ET AL. 30 3.41 (q, 1H, J = 7.1 Hz, rotamer2 CH2), 3.07 (s, 1.5H, rotamer1 CH3), 2.99 (s, 1.5H, rotamer2 CH3), 1.61 (s, 2H, NH2), 1.38 (d, 3H, J = 6.6 Hz, CH3), 1.24 (t, 1.5H, J = 6.1 Hz, rotamer1 CH3), 1.19 (t, 1.5H, J = 6.0 Hz, ro- tamer2 CH3); 13C-NMR (100MHz, CDCl3): δC 154.45 (d, J = 18.0 Hz, 1C, C=O), 151.57 (Caryl), 149.22 (Caryl), 129.10 (Caryl), 122.39 (Caryl), 120.02 (Caryl), 118.95 (Caryl), 50.95 (1C, CH), 43.94 (1C, CH2 of carbamoyl), 34.11 (rotamer1 Mecarbamoyl), 33.68 (rotamer2 Mecarbamoyl), 25.40 (1C, CH3), 13.10 (rotamer1 Mecarbamoyl), 12.35 (rotamer2 Mecarbamoyl); ESI-MS m/z (%): 206.0 (M-NH2, 100), 207.1 (M+ – NH2, 20), 223.1 (M+ + H, 15). The intermediate stages have also isolated for confir- mation by analytical and spectral data. 2-Methyl propane-2-sulfinic acid [1-(3-methoxy- phenyl)ethylidene]amide (19): yellow oil, yield 87%; [α]D 20 – 0.5˚ (c 1.0, CHCl3); HPLC Purity: 96.8%; IR (Neat, νmax, cm–1): 1089 (S-O), 1723 (C=O), 1575 (C=N), 2975 (aliphatic CH); 1H NMR (400MHz, CDCl3): δH 7.71 (d, 1H, J = 7.5 Hz, ArH), 7.59 (s, 1H, ArH), 7.40 (t, 1H, J = 7.9 Hz , ArH), 7.25 (d, 1H, J = 8.0 Hz, ArH), 3.48 (q, 1H, J = 7.0 Hz, rotamer1 CH2), 3.41 (q, 1H, J = 7.0 Hz, rotamer2 CH2), 3.08 (s, 1.5H, rotamer1 CH3), 2.99 (s, 1.5H, rotamer2 CH3), 2.74 (s, 3H, CH3), 1.30 (s, 9H, C(CH3)3), 1.24 (t, 1.5H, J = 7.0 Hz, rotamer1 CH3), 1.19 (t, 1.5H, J = 7.0 Hz, rotamer2 CH3); 13C NMR (100MHz, CDCl3): δC 175.27 (1C, C=N), 154.12 (d, J = 18.0 Hz, 1C, C=O), 151.52 (Caryl), 139.93 (Caryl), 129.11 (Caryl), 125.12 (Caryl), 123.96 (Caryl), 120.53 (Caryl), 57.55 (1C, Ct-butyl), 44.02 (1C, CH2 of carbamoyl), 34.19 (ro- tamer1 Mecarbamoyl), 33.76 (rotamer2 Mecarbamoyl), 22.46 (3C, Ct-butyl), 19.77 (1C, Me), 13.14 (rotamer1 Mecarbamoyl), 12.34 (rotamer2 Mecarbamoyl); ESI-MS m/z (%): 346.9 (M+ + Na, 100), 325.0 (M + H, 45), 306.9 (20), 291 (65), 250.9 (47), 243.0 (45). 2-Methyl propane-2-sulfinic acid [1-(3-methoxy- phenyl) ethyl]amide (20): yellow oil, yield 98%; [α]D 20 + 26.0˚ (c 1.0, CHCl3); Chiral HPLC Purity: 99.95%, Column: chiral cel AD-H 250 × 4.6 mm/5 µm (SRC- 581), mobile phase A: n-hexane:IPA:TFA (90:10:0.1), Iso: 100%, concentration: 10 mg/10 mL, diluent: ethanol, run time: 35 min, temperature: 25˚C, flow rate: 0.8 mL/min, UV: 215 nm, retention time: (S)-isomer: 12.78 min and (R)-isomer: 7.72 min; HPLC Purity: 92%; IR (Neat, νmax, cm–1): 1055 (S-O), 1718 (C = O), 2978 (ali- phatic CH), 3234 (NH); 1H NMR (400MHz, CDCl3): δH 7.32 (t, 1H, J = 7.8 Hz, ArH), 7.18 (d, 1H, J = 7.6 Hz , ArH), 7.08 (s, 1H, ArH), 7.04 (d, 1H, J = 7.0 Hz, ArH), 4.54 (q, 1H, J = 3.7 Hz, CH), 3.51 (s, 1H, NH), 3.47 (q, 1H, J = 7.0 Hz, rotamer1 CH2), 3.41 (q, 1H, J = 7.1 Hz, rotamer2 CH2), 3.06 (s, 1.5H, rotamer1 CH3), 2.98 (s, 1.5H, rotamer2 CH3), 1.50 (d, 3H, J = 6.5 Hz, CH3), 1.25 (t, 1.5H, J = 7.0 Hz, rotamer1 CH3), 1.20 (s, 9H, C(CH3)3), 1.19 (t, 1.5H, J = 7.0 Hz, rotamer2 CH3); 13C NMR (100MHz, CDCl3): δC 154.20 (d, J = 18.0 Hz, 1C, C=O), 151.59 (Caryl), 145.27 (Caryl), 129.39 (Caryl), 123.28 (Caryl), 121.12 (Caryl), 119.85 (Caryl), 55.33 (1C, Ct-butyl), 53.51 (1C, CH), 43.92 (1C, CH2 of carbamoyl), 34.08 (rotamer1 Mecarbamoyl), 33.68 (rotamer2 Mecarbamoyl), 29.14 (1C, Me), 22.48 (3C, Ct-butyl), 13.09 (rotamer1 Me- carbamoyl), 12.32 (rotamer2 Mecarbamoyl); ESI-MS m/z (%): 348.9 (M+ + Na, 90), 327.0 (M + H, 100), 293.0 (23), 252.9 (54), 245.0 (26), 194.0 (80). Route B. Synthesis of 2-Methyl propane-2-sulfinic acid [1-(3- hydroxyphenyl)ethyl]amide (23): 3-Hydroxyacetophenone (10.0 g, 0.066 mol) is re- fluxed with (S)-8 (8.07 g, 0.066 mol), titanium tetra eth- oxide (30.4 g, 0.133 mol) in THF (100 ml) for 30 hr. Cool to ambient temperature and further to –48 to –52˚C and added NaBH4 (3.8 g, 0.099 mol) in 4 portions. Stir for 3 hours to complete the reduction of imine intermedi- ate 22. Gradually warm to –5˚C and added methanol (10 ml) drop wise at –5˚C to 0˚C. Stir for 30 minutes, added ethyl acetate (50 ml) and water (100 ml). Stir for 30 minutes to warm to rt and filtered off the salts. Separate organic layer and aqueous layer is washed with ethyl acetate (50 ml). Combined organic layers are washed successively with water (2 × 50 mL) and brine solution (30 ml). The organic layer is dried over anhydrous Na2SO4 and evaporated to afford pale yellow solid of 23. Wt 13.7 g; yield 77.6%; mp 140˚C - 144˚C; [α]D 23 + 25.5˚ (c 1.0, MeOH); Chiral HPLC: 94.76%, Column: chiral pak AD-H 0.46cm × 25cm (SRC-581), mobile phase A: n-hexane:IPA:DEA (90:10:0.1), Iso: 100%, concentration: 50 mg/10mL, diluent: ethanol, run time: 45 min, temperature: 25˚C, flow rate: 1 mL/min, UV: 230nm, retention time: (S)-isomer: 24.87 min and (R)-isomer: 8.13 min; IR (KBr, cm–1): 3277 (NH), 3086 (OH), 2972 (aliphatic CH), 1017 (S-O); 1H NMR (400MHz, CDCl3): δH 8.2 (br s, 1H, OH), 7.15 (t, 1H, J = 8.0 Hz, ArH), 6.81 (d, 1H, J = 7.7 Hz, ArH), 6.77 (s, 1H, ArH), 6.76 (d, 1H, J = 7.5 Hz, ArH), δ 4.41 (q, 1H, J = 3.5 Hz, CH), δ 3.64 (s, 1H, NH), 1.46 (d, 3H, J = 6.5 Hz, CH3), 1.26 (s, 9H, C(CH3)3); 13C NMR (100MHz, DMSO-d6): δC 157.57 (1C, Caryl), 147.13 (1C, Caryl), 129.41 (1C, Caryl), 117.50 (1C, Caryl), 114.07 (1C, Caryl), 113.77 (1C, Caryl), 55.44 (1C, Ct-butyl), 54.47 (1C, CH), 24.50 (1C, CH3), 22.97 (3C, Ct-butyl); ESI-MS m/z (%): 242.4 (M + H, 100), 168.3 (50), 125.1 (10). The intermediate stages have also isolated for confir- mation by analytical and spectral data. 2-Methyl propane-2-sulfinic acid [1-(3-hydroxy- phenyl)ethylidene]amide (22): yellow crystals, yield 77.6%; mp 137˚C - 142˚C; [α]23 D + 3.0˚ (c 1.0, MeOH); Chiral HPLC purity: 99.98%, Column: chiral pak AD-H Copyright © 2011 SciRes. IJOC  V. R. ARAVA ET AL.31 0.46cm × 25cm (SRC-581), mobile phase A: n-hexane: IPA:DEA (90:10:0.1), Iso: 100%, concentration: 5 mg/10mL, diluent: ethanol, run time: 35 min, temperature: 25˚C, flow rate: 1 mL/min, UV: 254nm, retention time: (S)-isomer: 16.16 min and (R)-isomer: 12.55 min; IR (KBr, cm–1): 3180 (OH), 2987 (aliphatic CH), 1597 (C=N), 1028 (S-O); 1H NMR (400MHz, CDCl3): δH 7.72 (br s, 1H, OH), 7.43 (s, 1H, ArH), 7.32 (d, 1H, J = 7.4 Hz, ArH), 7.23 (t, 1H, J = 7.9 Hz, ArH), 7.01 (d, 1H, J = 7.9 Hz, ArH), 2.68 (s, 3H, CH3), 1.32 (s, 9H, C(CH3)3); 13C NMR (100MHz, DMSO-d6): δC 176.82 (1C, C=N), 157.78 (1C, Caryl), 140.01 (1C, Caryl), 129.98 (1C, Caryl), 119.34 (1C, Caryl), 118.52 (1C, Caryl), 113.92 (1C, Caryl), 57.08 (1C, Ct-butyl), 22.37 (3C, Ct-butyl), 20.03 (1C, CH3); ESI-MS m/z (%): 240.3 (M + H, 65), 184.2 (100), 166.1 (82). Synthesis of (S)-Ethyl methyl carbamic acid 3-(1- aminoethyl)phenyl ester (21): a mixture of 23 (13.0 g, 0.053 mol), K2CO3 (14.9 g, 0.107 mol) and N-ethyl- N-methyl carbamoyl chloride (8.18 g, 0.067 mol) in ethyl acetate (130 ml) is refluxed for 4 hours. Cool to room temperature and washed with water (3 × 130 mL), dried over anhydrous Na2SO4, filtered and evaporated under reduced pressure to get pale yellow oil of 20 (17.37 g, 99% yield), which is hydrolyzed with 13% methanolic HCl (18 ml) and then alkali treatment with aq.NaOH at rt, extraction into ethyl acetate and evaporation of the sol- vent under reduced pressure afford pale yellow oil of 21. Wt 11.25 g, yield 95%. Synthesis of (S)-Rivastig mine (1): the pale yellow oil 21 (65.0 g, 0.43 mol) is dissolved in formic acid (198.0 g, 4.3 mol), formaldehyde (44.0 g, 3.4 mol (35% solution in water) and water (650 ml). Heated to reflux temperature (95˚C - 100˚C) and stirred for 5 hr. Cool to ambient temperature; wash with ethyl acetate (2 × 130 ml) to re- move the impurities. Aqueous layer is separated, ad- justed pH to 10 with 20% aq. NaOH solution (758 ml) and extracted with ethyl acetate (2 × 325 ml). The com- bined organic layers are washed with brine solution (100 ml), dried over anhydrous sodium sulfate and evaporated under reduced pressure at 45˚C - 50˚C to get pale yellow oil. Wt 78.4 g; yield 95%; [α]D 20 – 35.2˚ (c 1.0, CHCl3) (lit [6]: [α]D 20 – 33.9˚ (c 1.0, CHCl3); Chiral HPLC Pu- rity 100%, Column: chiral pak OD-H 250 × 4.6 mm, mobile phase A: n-hexane:IPA:TFA (80:20:0.1), Iso: 100%, concentration: 10 mg/25mL, diluent: ethanol, run time: 25 minutes, temperature: 25˚C, flow rate: 0.8 mL/min, UV: 215 nm, retention time: (S)-isomer: 14.41 min and (R)-isomer: 9.54 min;; IR (Neat, νmax, cm–1): 2975 (aliphatic CH), 1725 (C=O); 1H NMR (400MHz, CDCl3): δH 7.28 (t, 1H, J = 7.8 Hz, ArH), 7.10 (d, 1H, J = 7.8 Hz, ArH), 7.05 (s, 1H, ArH), 7.00 (d, 1H, J = 7.8 Hz, ArH), 3.44 (q, 1H, J = 7.5 Hz, CH2 rotamer 1), 3.41 (q, 1H, J = 7.5 Hz, CH2 rotamer 2), 3.23 (q, 1H, J = 6.6 Hz, –CH), 3.05 (s, 1.5H, CH3 rotamer 1), 2.97 (s, 1.5H, CH3 rotamer 2), 2.19 (s, 6H, N(CH3)2), 1.34 (d, 3H, J = 6.7 Hz, CH3), 1.23 (t, 1.5H, J = 7.2 Hz, CH3 rotamer 1), 1.17 (t, 1.5H, J = 7.2 Hz, CH3 rotamer 2) ; 13C NMR (100 MHz, CDCl3): δC 154.41 (d, J = 18.0 Hz, 1C, C = O), 151.41 (Caryl), 145.63 (Caryl), 128.77 (Caryl), 124.11 (Caryl), 120.60 (Caryl), 120.14 (Caryl), 65.53 (1C, CH), 43.92 (1C, CH2 of carbamoyl), 43.11, 34.09 (rotamer1 Mecarbamoyl), 33.67 (rotamer2 Mecarbamoyl), 19.98 (1C, Me), 13.12 (rotamer1 Mecarbamoyl), 12.37 (rotamer2 Mecarbamoyl); ESI-MS m/z (%): 251.2 (M + H, 100), 206.2 (60). Preparation of (S)-(+)-rivastigmine hydrogentar- tarate (1): (S)-(-)-rivastigmine 1 (4.5 g) and L-(+)- tartaric acid (2.7 g) are dissolved in anhydrous ethanol (12.5 mL) at 60˚C. Ethyl acetate (63 mL) is added at the same temperature and stirred for 10 minutes. Cool the solution to room temperature and to crystallize at +5˚C for a period of 12 hours. The precipitated solid is col- lected through filtration and washed the cake with ethyl acetate (10 mL). Dry the solid at 50˚C under vacuum to afford 6.7 g of the desired product. Yield 92%; [α]D 20 + 4.6˚ (c 5.0, ethanol); mp 117˚C - 120˚C; Chiral HPLC Purity: 100% ee. 4. Conclusions In summary, a high yielding stereoselective and short synthesis of (S)-Rivastigmine is described in two ways with an overall isolated yield of 76% and 70% respec- tively via 3 step procedure starting from 3-hydroxyaceto- phenone, a commercially available intermediate. [23] 5. Acknowledgements We greatly acknowledge our thanks to Mr. Venkat Jasti, CEO for his excellent support, Mr. K. R. S. Sharma for Chiral HPLC method development and the analytical group of SUVEN for providing spectral data. 6. References [1] N. C. Berchtold and C. W. Cotman, “Evolution in the Conceptualization of Dementia and Alzheimer’s Disease: Greco-Roman Period to the 1960s,” Neurobiology of Ag- ing. Vol. 19, No. 3, 1998, pp. 173-189. doi:10.1016/S0197-4580(98)00052-9 [2] J. Corey-Bloom, R. Anand and J. Veach, “A Randomized trial Evaluating the Efficacy and Safety of ENA 713 (Rivastigmine Tartarate), a New Acetylcholinesterase In- hibitor, in Patients with Mild to Moderately Severe Alz- heimer’s Disease,” International Journal of Geriatric Psychopharmacology, Vol. 1, No. 2, 1998, pp. 55-65. [3] M. Rosler, R. Anand, A. Cicin-Sain, S. Gauthier, Y. Agid, P. Dal-Bianco, H. B. Stähelin, R. Hartman and M. Copyright © 2011 SciRes. IJOC  V. R. ARAVA ET AL. Copyright © 2011 SciRes. IJOC 32 Gharabawi, “Efficacy and Safety of Rivastigmine in Pa- tients with Alzheimer’s Disease: International Random- ized Controlled Trial,” British Medical Journal, Vol. 318, No. 7184, 1999, pp. 633-640. [4] S. I. Finkel, “Effects of Rivastigmine on Behavioral and Psychological Symptoms of Dementia in Alzheimer’s Disease,” Clinic Therapeutics, Vol. 26, No. 7, 2004, pp. 980-990. doi:10.1016/S0149-2918(04)90172-5 [5] M. Rosler, W. Retz, P. Retz-Junginger and H. J. Dennler, “Effects of Two-Year Treatment with the Cholinesterase Inhibitor Rivastigmine on Behavioural Symptoms in Alz- heimer’s Disease,” Behavioral Neurology, Vol. 11, No. 4, 1998, pp. 211-216. [6] M. Emre, D. Aarsland and A. Albanese, “Rivastigmine for Dementia Associated with Parkinson’s Disease,” The New England Journal of Medicine, Vol. 315, 2004, pp. 2509-2518. doi:10.1056/NEJMoa041470 [7] C. Gabelli, “Rivastigmine: An Update on Therapeutic Efficacy in Alzheimer’s Disease and Other Conditions,” Current Medical Research & Opinion, Vol. 19, No. 2, 2003, pp. 69-82. [8] M. Wakayama and J. A. Ellman, “Recycling the Tert-Butanesulfinyl Group in the Synthesis of Amines Using Tert-Butanesulfinamide,” Journal of Organic Chemistry, Vol. 74, No. 4, 2009, pp. 2646-2650. doi:10.1021/jo9001883 [9] M. T. Robak, M. A. Herbage and J. A. Ellman, “Synthe- sis and Applications of Tert-Butanesulfinamide,” Chemical Reviews, Vol. 110, No. 6, 2010, pp. 3600-3740. doi:10.1021/cr900382t [10] A. V. Reddy, S. U. B. Rao, G. L. Narasimha and P. K. Dubey, “Improved Process for the Preparation of Tamsu- losin Hydrochloride,” Synthetic Communications, Vol. 39, No. 8, 2009, pp. 1451-1456. doi:10.1080/00397910802519216 [11] A. Gaitonde, M. Mangle and S. R. Pawar, “Novel Process for the Preparation of Aminoalkyl Phenyl Carbamates,” Chemical Abstracts, Vol. 143, 2005, Article ID: 77963. [12] H. Stepankova, J. Hajicek and S. Simek, “A Method of Production of (-)-(S)-3-[1-(Dimethylamino)ethyl] phenyl- N-ethyl-N-methylcarbamate,” Chemical Abstracts, Vol. 142, 2004, Article ID: 6315. [13] M. J. V. Garrido, A. M. Montserrat and M. J. Juarez, “Method of Obtaining Phenyl Carbamates,” 2007, Chemical Abstracts, Vol. 146, 2007, Article ID: 206113. [14] J. Feng, W.-M. Chen and P.-H. Sun, “Synthesis of S-(+)-Rivastigmine Hydrogen Tartarate,” Journal of Southern Medical University, Vol. 27, No. 2, 2007, pp. 177-180. [15] A. A. Boezio, J. Pytkowicz, A. Cote and A. B. Charette, “Asymmetric, Catalytic Synthesis of α-Chiral Amines Using a Novel Bis(Phosphine)Monoxide Chiral Ligand,” Journal of the American Chemical Society, Vol. 125, No. 47, 2003, pp. 14260-14261. doi:10.1021/ja038291+ [16] M. Hu, F. L. Zhang and M. H. Xie, “Novel Convenient Synthesis of Rivastigmine,” Synthetic Communications, Vol. 39, No. 9, 2009, pp. 1527-1533. doi:10.1080/00397910802531948 [17] J. M. Sanchez, M. R. Mata, E. Busto, V. G. Fernandez and V. Gotor, “Chemoenzymatic Synthesis of Rivastig- mine Based on Lipase-Catalyzed Processes,” The Journal of Organic Chemistry, Vol. 74, No. 15, 2009, pp. 5304-5310. doi:10.1021/jo900784g [18] K. Han, C. Kim, J. Park and M.-J. Feng, “Chemoenzy- matic Synthesis of Rivastigmine via Dynamic Kinetic Resolution as a Key Step,” The Journal of Organic Chemistry, Vol. 75, No. 9, 2010, pp. 3105-3108. doi:10.1021/jo9027374 [19] K. A. Kumar, M. A. Reddy, T. S. Kumar, B. V. Kumar, K. B. Chandrasekhar, P. R. Kumar and M. Pal, “Achiral Bis-Imine in Combination with CoCl2: A Remarkable Effect on Enantioselectivity of Lipase-Mediated Acetyla- tion of Racemic Secondary Alcohol,” Beilstein Journal of Organic Chemistry, Vol. 6, 2010, pp. 1174-1179. doi:10.3762/bjoc.6.134 [20] M. Fuchs, D. Koszelewski, K. Tauber, W. Kroutil and K. Faber, “Chemoenzymatic Asymmetric Total Synthesis of (S)-Rivastigmine Using ω-Transaminases,” Chemical Communications, Vol. 46, No. 30, 2010, pp. 5500-5502. doi:10.1039/c0cc00585a [21] D. Guijarro, O. Pablo and M. Yus, “Asymmetric Synthe- sis of Chiral Primary Amines by Transfer Hydrogenation of N-(tert-Butanesulfinyl)ketimines,” The Journal of Or- ganic Chemistry, Vol. 75, No. 15, 2010, pp. 5265-5270. doi:10.1021/jo101057s [22] A. V. Reddy, G. Laxminarasimhulu and P. K. Dubey, “Facile Enantioselective Synthesis of (S)-Rivastigmine & (+)-NPS-R-568 a Calcimimetic Agent,” Der Pharma Chemica, Vol. 3, No. 1, 2011, pp. 426-433. [23] A. V. Reddy, “Developing Chemical Processes for Active Pharmaceutical Ingredients (API’s),” 3rd International Conference and Exhibition, Mumbai, 10-11 January 2011, Abstract No. 3.
|