M. K. Haldar et al.
2. Experimental
Natural Indian magnesite of Salem district in Tamilnadu (supplied by erstwhile Burn Standard Co. Ltd., pre-
sently SAIL Refractory Co. Ltd, India) was selected for the present study. The CaCO3 of S.D. Fine Chemical
Limited, India was selected for changing the CaO/SiO2 ratio of the batches. The chemical analysis of the magne-
site, as received, is delineated in Table 1. The analysis was done by standard wet chemical procedure. 0.25 g of
the solid powdered sample was taken into a platinum crucible and the sample was thoroughly mixed with borax
sodium carbonate fusion mixture and melted at around 900˚C for 1h. After melting, the mass was cooled down
and digested with nitric acid to obtain a solution. Fe2O3 and TiO2 were measured by colorimetric method. MgO,
Al2O3, CaO were determined by complexometric method using EDTA solution. Na2O and K2O were measured
by flame photometry using hydrofluoric acid and perchloric acid digested magnesite solution. SiO2 was meas-
ured by gravimetric method.
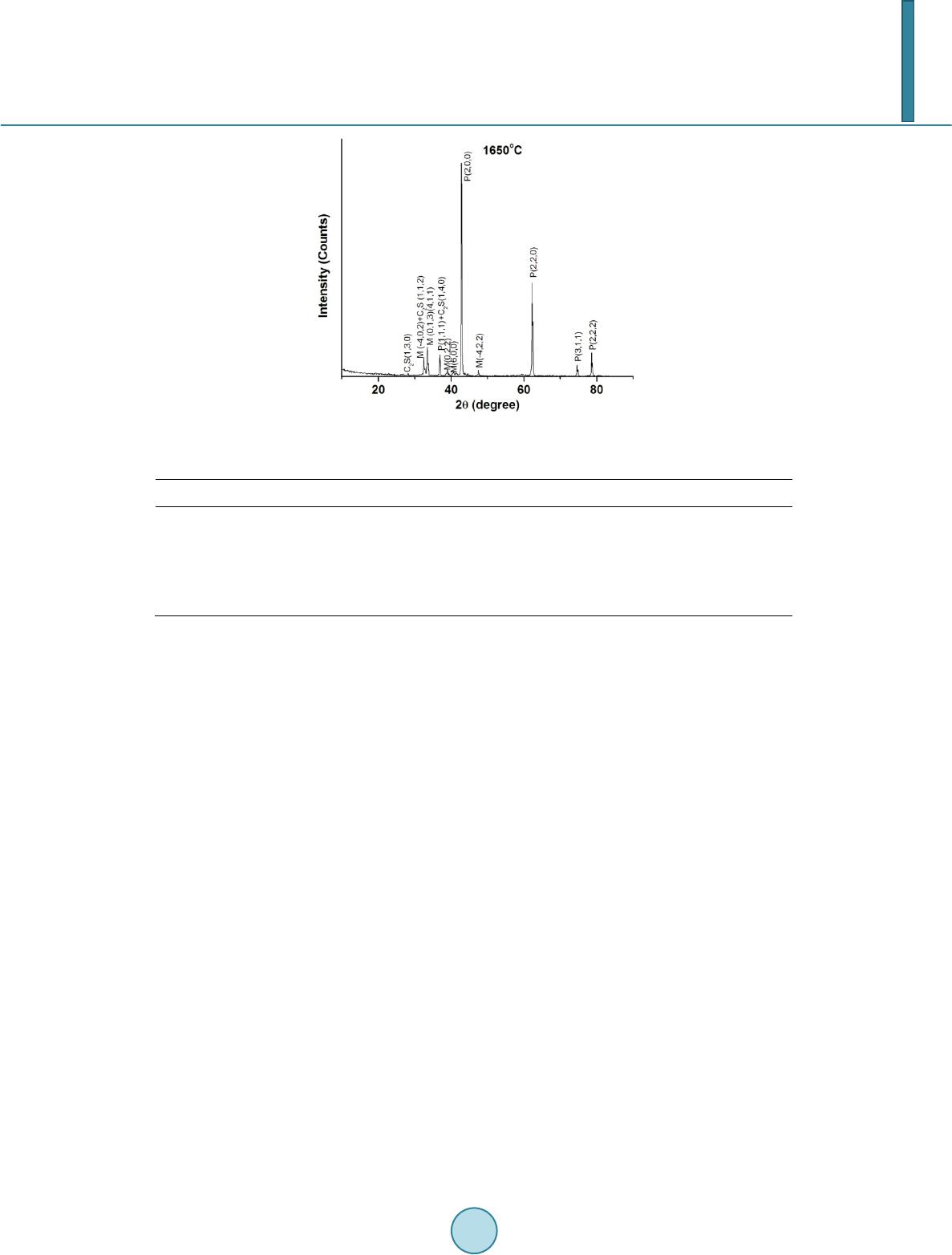
The structural characterization of the sintered aggregates in terms of phase identification and evaluation of
crystal structure parameters like crystallite size, lattice strain, lattice parameters, unit cell volume etc was per-
formed by X-ray diffraction technique. The XRD patterns of the samples were recorded in X’pert Pro MPD dif-
fractometer (PANalytical) by X’Celerator operating at 40 kV and 30 mA, using Ni-filtered CuKα radiation. The
XRD data were recorded with step size of 0.05˚ (2θ), step time of 75 sec, from 10˚ to 90˚. The magnesite ores
were crushed in a jaw crusher and hammered to pass through 100 mesh sieve. These powders were mixed with
CaCO3 to have a mole ratio of CaO:SiO2 = 2:1. The batch was attrition milled in attritor mill with isopropyl al-
cohol for 1h. The slurry thus obtained was dried at 50˚C ± 2˚C in a laboratory oven for 2 h and passed through
100 mesh sieve to get the desired powder. Powdered sample was mixed with 5% (w/v) of polyvinyl alcohol so-
lution as binder and subsequently granulated by sieving and uni-axially pressed at 140 MPa pressure into pellets
of 2.5 cm dia and 1 cm height for densification studies and bars of 80 mm × 8 mm × 8 mm for the rmo-mechan-
ical and mechanical studies and few bars were cold iso-statically pressed at 140 MPa for thermal shock studies.
All the pressed samples were dried at 110˚C ± 5˚C and sintered in the temperature range of 1550˚C - 1700˚C
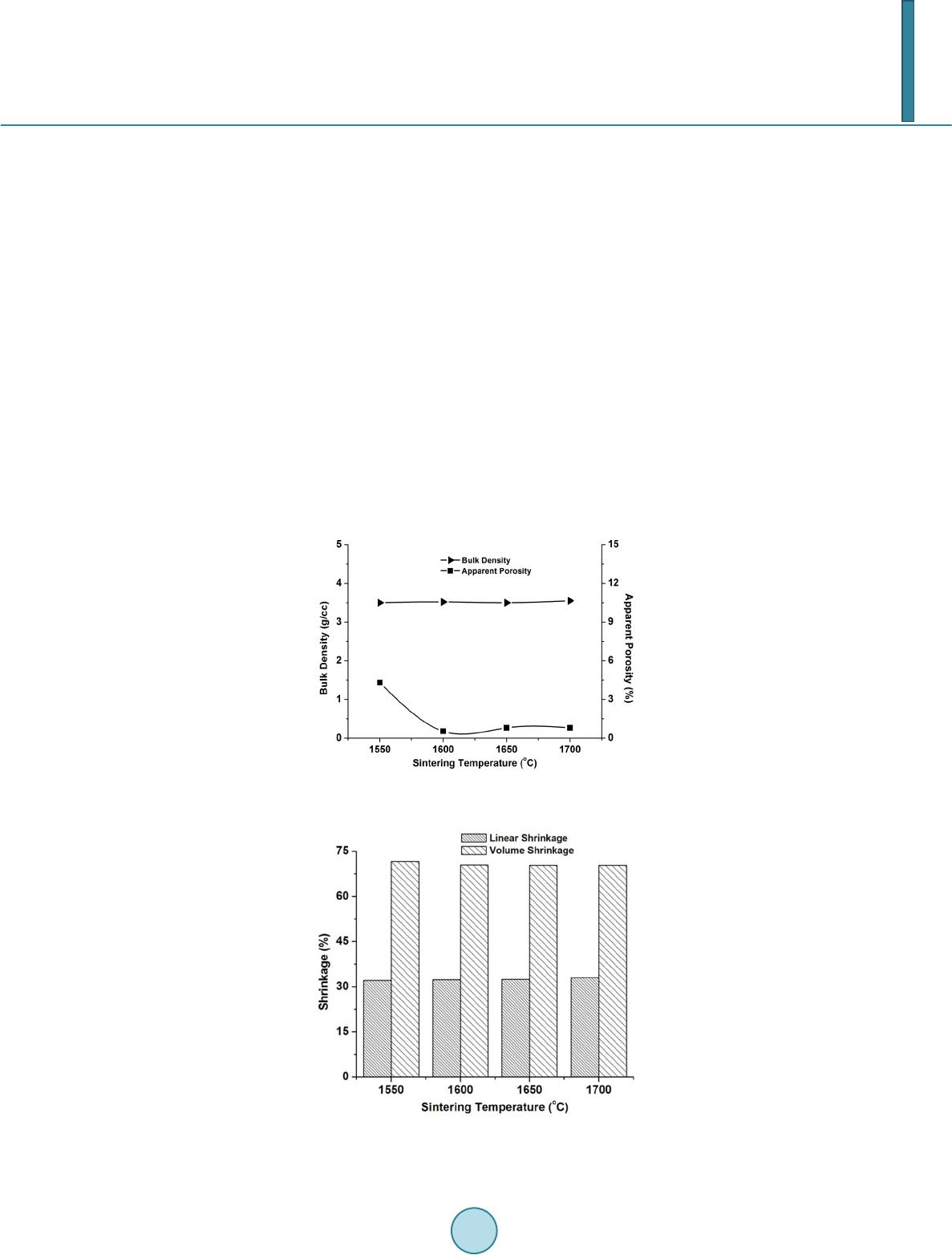
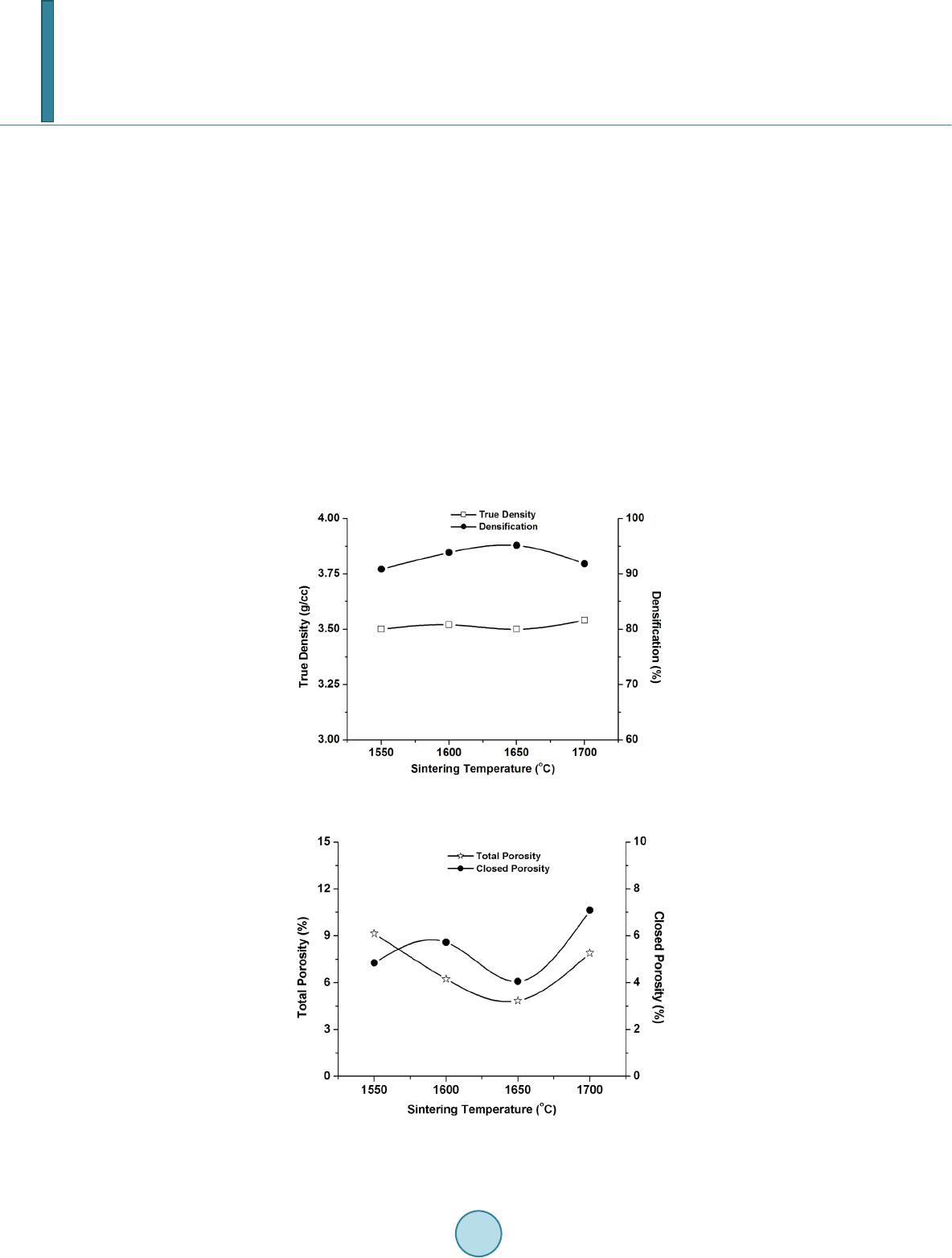
with 2 h of soaking at the peak temperature. Sintered samples were characterized in terms of bulk density, ap-
parent porosity, closed porosity, true density, relative density (percentage densification). The bulk density and
apparent porosity were measured by water displacement method using Archimedes’ principle. True density was
measured with powdered samples in a pycnometer bottle. From these two values, percentage densification of the
sintered samples was measured. Closed porosity was measured by subtracting apparent porosity values from to-
tal porosity. The linear shrinkage of the sintered samples was measured using digital slide calipers. Crystalline
phases of the sintered samples were identified from XRD patterns. Percentages of crystalline phases were esti-
mated for the samples from X-ray diffraction line profile analysis using Rietveld method [1] by X’Pert High
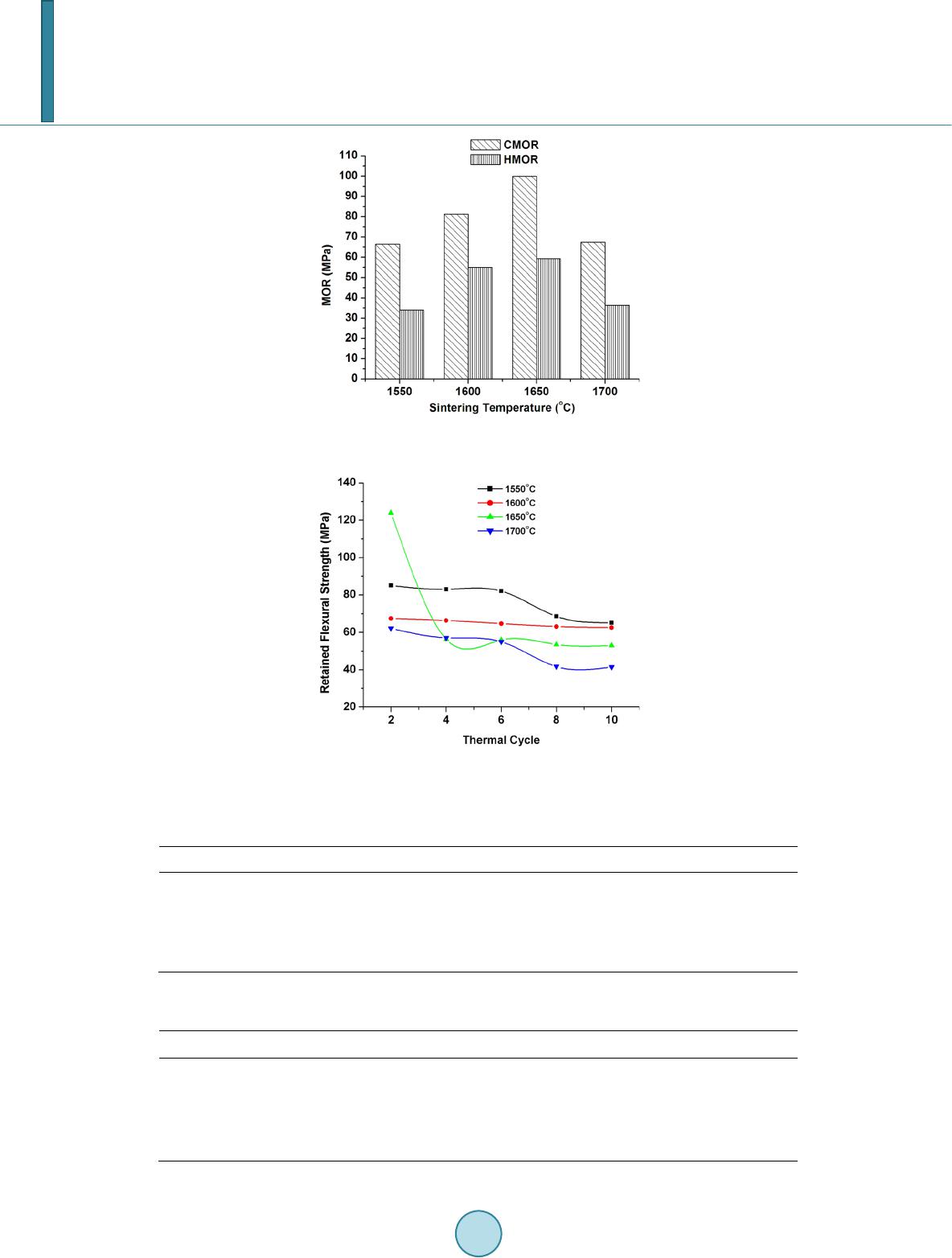
Score Plus software (PANalytical). The flexural strength at room temperature (cold MOR) of the sintered rec-
tangular bars was measured by three point bending method using an Instron-1185 universal testing machine. The
span of the bars was 40 mm and the cross head speed was 0.5 mm∙min−1. The high temperature modulus of rup-
ture strength was measured at 1300˚C by an instrument supplied by Stedfast International Company Limited,
Table 1. Chemical analysis of as received magnesite.
Constituents Wt% Wt% (loss free basis)
SiO2 2.44 4.8
Al2O3 0.17 0.34
Fe2O3 0.24 0.48
TiO2 0.02 0.4
CaO 1.41 2.79
MgO 45. 68 90.49
Na2O 0.08 0.16
K2O 0.05 0.99
L.O.I. 49.52 -