Natural Science
Vol.6 No.2(2014), Article ID:43144,12 pages DOI:10.4236/ns.2014.62010
The role of polysaccharides in the molecular evolution of biopolymers
1Research and Production Association “Plazan”, Moscow, Russia; *Corresponding Author: zanol@yandex.ru
2Bashkir State University, Ufa, Russia
3Institute of Organic Chemistry, Russian Academy of Sciences, Ufa, Russia
4Biochemistry and Genetics, Institute of Russian Academy of Sciences, Ufa, Russia
Copyright © 2014 Aleksander N. Zimnitskii et al. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited. In accordance of the Creative Commons Attribution License all Copyrights © 2014 are reserved for SCIRP and the owner of the intellectual property Aleksander N. Zimnitskii et al. All Copyright © 2014 are guarded by law and by SCIRP as a guardian.
Received 21 December 2013; revised 21 January 2014; accepted 29 January 2014
Keywords: Molecular Evolution; Aminoacylation; Polysaccharides; Amino Acids; Nucleic Acids; Guantum Chemistry; PCR
ABSTRACT
The empirical (biochemical, the PCR method) and computational (quantum-chemical, PM3) methods have shown the ability of polysaccharides to catalyze the polymerization of amino acids and nucleotides within the physiologically acceptable temperature range. The possibility of nucleotide aminoacylation in the presence of polysaccharides has been established. The suggestion has been made that abiogenic aminoacylation of nucleotides by polysaccharides served as the prototype of the original aminoacylpre-tRNA-synthetase activity and subsequently determined the formation of the modern mechanism of genetic information transfer via three biopolymer types—nucleic acids, proteins and polysaccharides.
1. INTRODUCTION
Chemical and molecular evolution of biopolymer monomers—proteins and nucleic acids (NA) is the fundamental issue in the understanding of life origin on our planet. The renowned works by S. Miller [1] demonstrated the possibility in principal of nucleic acid (NA) formation from the simplest chemical compounds—methane, ammonia, hydrogen and water vapor. Furthermore, the research conducted by the academician V. N. Parmon
[2] made it possible to prove the possibility of practically selective ribose and simplest sugar synthesis from formaldehyde in the presence of phosphate anions (natural apatite). Abiogenic formation of the monomers of modern biopolymers was verified not only by the model chemical experiments, but also by the study of interstellar and alien matter composition [3]. Thus, today it is beyond any doubt that amino acids, sugars and nucleotides were formed abiogenically at the initial stages of the evolution both inside and outside the primitive atmosphere of the Earth. Besides, S. W. Fox [4] showed experimentally that proteinaceous multi-molecule systems can be formed under achievable in the natural conditions thermal influence on amino acid mixture. The so-called thermal proteinoids of S. W. Fox and oligonucleotides of L. Orgel were effectively formed at temperatures above 100˚C. These processes were, thus, localized only in the volcanic activity zone in the conditions of the Earth’s primitive atmosphere, which led to the destruction of polymers under the influence of the excessive thermal energy. At the same time, a great number of scientists [6] believe that life origin on our planet is connected with the period of its cooling to temperatures below 100˚C and the formation of primary hydrosphere elements. Therefore, high temperatures apparently could not play the leading role in the system of biomolecule polymerization. A reaction catalyst is needed to promote this process in the physiological temperature range in the absence of excessive thermal energy. This catalyst should not only promote polymer assembly, but also have an information intensive structure, which is in its turn able to change and evolve. Clays which, according to J. Bernal [7], could have played this part, are not able to function as catalysts of monomer polymerization, despite their ability to adsorb biopolymers. Moreover, the ability of these minerals to evolve is out of the question, even taking into account the great variety of their crystal forms. Therefore, the search of the catalyst of the polymerization process of biologically significant monomers in the physiological temperature range is a vital issue in the understanding of the initial stages of biopolymer molecular evolution.
As discussed earlier, it is generally agreed that biomolecules were represented by sugars, nucleotides as well as amino acids at the initial stages of evolution [1-7]. According to the academician V. N. Parmon [2], sugar synthesis was more advantageous, since only sugars (as opposed to NA and amino acid (AA) possess the autocatalytic synthesis system working successfully in highly diluted solutions (the “formose” reaction of Butler, sugar synthesis from formaldehyde in the presence of calcium or magnesium ions). Moreover, it is well-known that calcium and magnesium ions form a complex with a sugar molecule to which a formaldehyde molecule is then joined. The formation of complexes from several sugar molecules was observed [2], suggesting the possibility of abiogenic oligoand polysaccharide (PS) formation in aqueous media. Thus, oligosaccharide appearance in the conditions of Earth’s primary atmosphere was more likely than oligopeptide or oligonucleotide appearance. Taking into account that a number of researchers earlier established the information capacity (microheterogeneity) of a polysaccharide chain [8-10], while we established the complementarity of NA to PS [11-14], the aim of the present work was to reveal the ability of PS to accelerate oligopeptide and oligonucleotide polymerization processes abiogenically.
2. MATERIALS AND METHODS
2.1. Quantum-Chemical Calculations
Theoretical calculations for the formation of nucleotide-polysaccharides (PS) complexes in this work were carried out by the PM3 semiempirical method [15] with the complete optimization of geometrical parameters employing the GAMESS program [16]. A global minimum of the total electron energy was sought by the Newton-Raphson method with an energy gradient of 0.1 kcal/mol ΔЕ, starting from various initial approximations of the complex structure.
2.2. Methods of Work with Amino Acids, Peptides and Polysaccharides
PS-AA complex was formed by dissolving 100 mg of PS (amylose—Am or polyuronides—PU) in 400 ml of water and adding 20 biogenic amino acids, 0.05 mm each. The mixture was made up to 500 ml with water. The solution was incubated at 40˚C for 1 hour, and then it was evaporated to 50 ml in vacuum. The complexes were precipitated by adding triple volume of ethanol at +4˚C for 12 hours. The precipitate was collected by centrifugation and dried out. The quantity of AA was determined in the precipitate.
The amino acid composition was determined by dissolving the AA-PS precipitates in 0.1 N НСl. The obtained solution was filtered, and AA were determined in the filtrate by a standard method on the Automatic Amino Acid Analyzer T-339 (Czech Republic). For this purpose, 100 µ of the sample was introduced by a sample loop onto a 31.5 cm high ion-exchange column containing ion-exchange resin LG ANB Ostion LB. Then a stepwise elution was conducted with рН 3.15, рН 5.25 and рН 9.25 buffered solutions. Acidic AA were eluted with the first buffer, neutral AA—with the second one, and basic AA—with the third one. AA were quantified photometrically after the ninhydrin reaction in a capillary reactor. The peaks were identified and quantified with reference to a standard AA mixture containing 2.5 mM/ ml of each amino acid. Assay sensitivity was 0.05 nM AA. PS mixtures precipitated by ethanol without AA served as the control.
The abiogenic formation of peptide bonds under thermal influence was carried out in the following way. 20 amino acids, 0.05 mM each, were dissolved in 50 ml of distilled water containing 100 mg of PS (Am or PU). The samples were evaporated to dryness in vacuum and the obtained crystals were exposed to different temperatures (20˚C, 40˚C, 60˚C, 120˚C, 130˚C) for 6 hours. The mixture of AA-PS crystals was then dissolved in water. The presence of peptide bonds was determined by the biuret method [17]. The control was amino acid mixture dissolved in 50 ml of distilled water without PS treated in the same way as the test sample. Amino acids were obtained from “Fluka” company.
2.3. Methods of Work with Nucleotides, Polynucleotides and Polysaccharides
As a rule, acidic polysaccharides and polyuronides in plants are represented by the monomers of two hexuronic acids—D-glucuronic acid and D-galacturonic acid. Dglucuronic acid is common in hemicellulose polysaccharides, whereas D-galacturonic acid in the form of galacturonans is the main component of pectic substances. It is worth noting that these polysaccharides can be easily obtained from oat seeds and apple pulp. Thus, acidic oligosaccharides were extracted from apples according to a standard method described earlier for oat seeds [11-14]. The extracted PU was additionally treated with deoxyribonuclease and proteases to prevent contamination with endogenous DNA. The enzymes from PU samples were then eliminated by three-fold treatment with phenol-chloroform mixture followed by three-fold precipitation by triple volume cooled ethanol [18].
Human placenta glycosaminoglycans were obtained by a standard method [11-14]. The purity of PS used in the experiment was controlled by electrophoresis on cellulose acetate bands in 0.1 N HCl.
5' nucleotide monophosphates (sodium salt) in ribo-, deoxyribo-form and oligo (dT)-cellulose were obtained from “Sigma-Aldrich”, amylose (neutral polysaccharide, NS) was obtained from “Serva Feinbiochemica GMBH”.
The procedure of adenosine monophosphate (AMP) polymerization (sodium salt) was as follows. 10 mg of AMP and 10 mg of PU were dissolved in 50 ml of H2O. Then the solution was evaporated to dryness in vacuum at a temperature of 40˚C and the obtained nucleotide PS crystals were exposed to different temperatures (20˚C, 40˚C, 60˚C, 120˚C, 130˚C) for 6 hours. Poly-A fragments of NA were extracted on oligo (dT)-cellulose by a standard method [19].
Deoxynucleotide polymerization in the presence of polyuronides (PU) was carried out by dissolving 10 mg of PU in 40 ml of water and adding 2.5 mg of each of the four nucleotides. The mixture was made up to 50 ml with H2O. The solution was incubated at 40˚C or 60˚C for 1 hour, then it was evaporated to dryness in vacuum and dissolved in 5 ml of water.
The complexes were precipitated by adding triple volume of ethanol at +4˚C for 12 hours. The precipitate was collected by centrifugation and dried out. The quantity of nucleotides was estimated spectrophotometrically by measuring the absorbance at 260 nm. The correspondence of the absorbance to the quantity of nucleotides was determined by a calibration curve individually for each nucleotide. PS alcohol precipitation in the absence of nucleotides served as the control.
Polymerase chain reaction (PCR) was conducted with short oligonucleotides in non-specific conditions of hybridization (the DNA-RAPD method). The amplification process was preceded by a preliminary denaturation at 94˚C for 2 min and consisted of 40 cycles, each one including the following steps: denaturation at 94˚C for 20 sec, annealing of the primers at 30˚C for 20 sec, and elongation at 72˚C for 20 sec. The following oligonucleotides were used for DNA-amplification: (AFK-3— acggtggacg, AFK-1—gcgtccaattc, ADE-1—tcgaatagc).
2.4. Gel Filtration
Gel-filtration on Sephadex G-25 was carried out on a column 30 cm long, 1.5 cm in diameter. The volume of the applied sample was 5 ml, fraction size was 3 ml. PU was detected spectrophtometrically by measuring the absorbance at 220 nm, whereas NA was detected by measuring the absorbance at 260 nm.
3. RESULTS
3.1. AA Polymerization on PS
The purity of PS used in the experiment was determined by biochemical analysis and was confirmed by electrophoresis. The results of the research are shown in Figure 1 and Table 1.
The formulas of PS used in the experiments can be represented as a recurrent dimer with the sequence AA, AB, BA, BB, where A is neutral sugar (hydroxymethyl group) and B is acidic sugar (carboxyl group). Bonding ability between amino acid radicals and monosaccharides was analyzed using the quantum-chemical method. The results are shown in Table 2.
As Table 2 demonstrates, end groups of amino acid radicals are able to form quite strong hydrogen bonds both with carboxyl and hydroxymethyl groups of monosaccharides. The energies of bonds between AA radical end groups and monosaccharides are not equal (Table 2). Five amino acids (Leu, Ile, Pro, Val, Ala, Gly) have practically zero bond energy between the radical and functional sugar groups. Three amino acids (Cys, Phe, Trp) are characterizes by a weak bond energy, 0 to 2 kcal/mol. Four amino acids (Tyr, Met, Thr, Ser) have a medium bond energy, 2 to 5 kcal/mol, whereas seven amino acids (Arg, Lys, Gln, Glu, Asp, Asn, His) have a high bond energy with the interaction strength at the carboxyl group being 5 to 10 kcal/mol.
It is worth noting that the bond energy between water
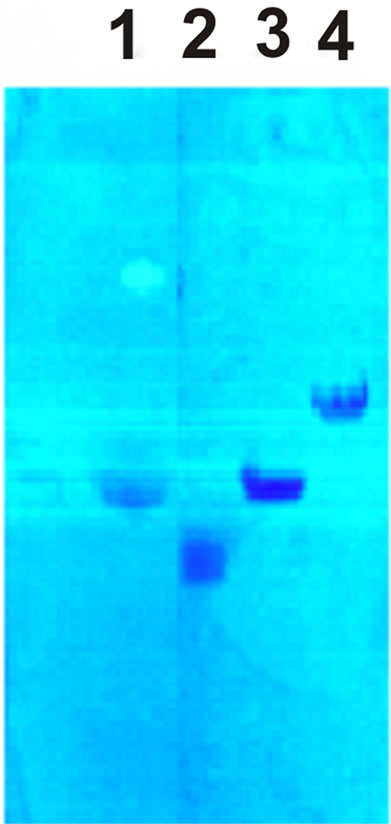
Figure 1. PS electrophoregram on cellulose acetate bands in 0.1 N HCl. 1—polyuronides, 2—heparin sulfate, 3—chondroitin sulfate, 4— hyaluronic acid.
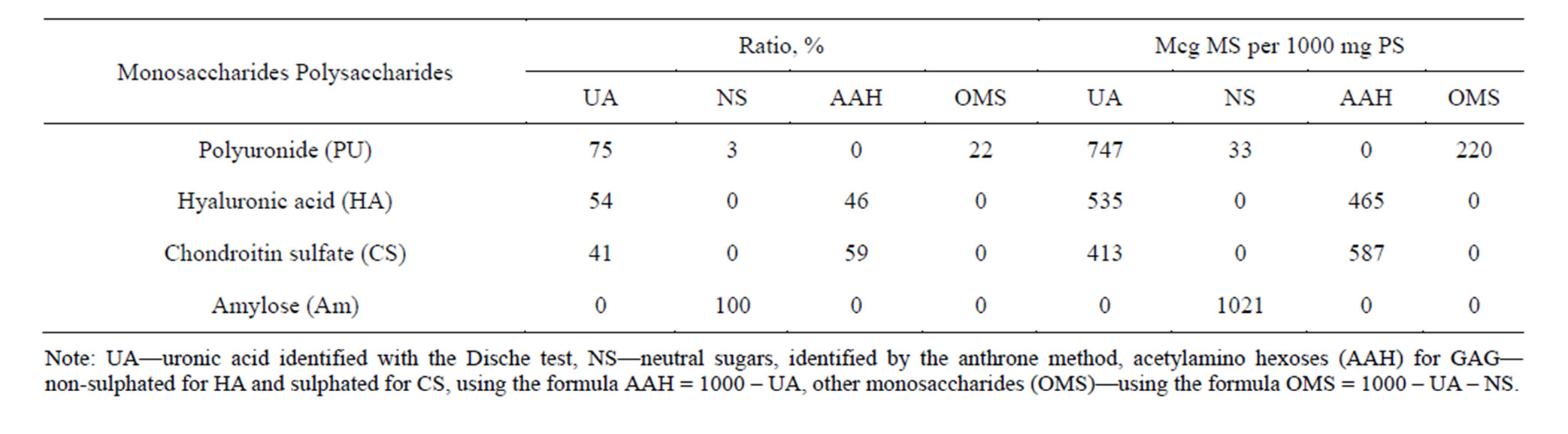
Table 1. Characteristics of PS used in the experiments.
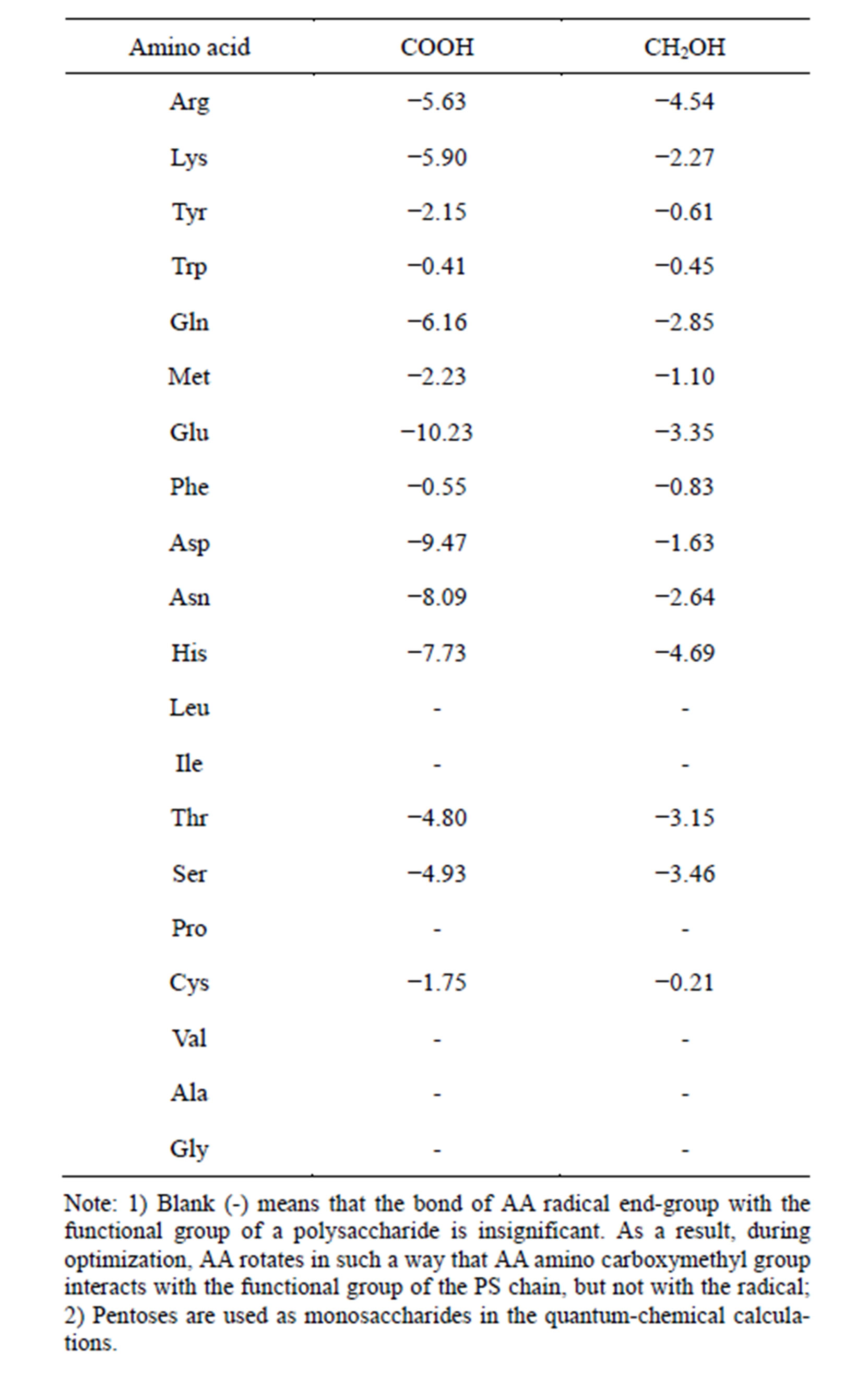
Table 2. Bond energy of the end-group of L-amino acid radical with the carboxyl or hydroxymethyl group of saccharides (∆Е, kcal/mol). Quantum-chemical calculation, the PM3 method.
and the carboxyl and hydroxymethyl groups of saccharides is −5.72 and −3.57 kcal/mol, respectively. In view of this fact the estimates of AA and PS radical bonds are more accurate during the dehydration of PS-AA complexes. Furthermore, aminocarboxymethyl groups of amino acids, themselves, are capable of forming bonds with the carboxyl and hydroxymethyl saccharide groups, ∆Е of which are −18.33 and −5.72 kcal/mol, respectively. These values are significantly higher than those of the bond energies formed in aqueous medium. Thus, PS are able to form hydrogen bonds with AA in aqueous medium and, at the same time, position the carboxyl group of the AA part in an optimal way in relation to the amino group of another AA to form random linear peptide bonds in the process of dehydration. These calculations testify that in solution the bulk of amino acids are bound to PS by their aminocarboxymethyl group. For some AA (two last classes), however, the bond energy of radical end-group is quite high. In this case the aminocarboxymethyl group of the given AA remains free, at a certain distance from the PS chain.
In order to verify the bonding ability between polysaccharides and amino acids, an experiment was conducted on polysaccharides in the solutions of amino acids present in equal proportions. It will be recalled that amylose is a homoglycan of monosachharides with the hydroxymethyl group (neutral polysaccharide, NS) and polyuronide is mainly a homoglycan of polysaccharides with the carboxyl group (acidic polysaccharide, AS). In fairly concentrated PS and amino acid solutions, PS monomers must bind with AA, if such a process exists. The subsequent ethanol precipitation of PS insolubilizes only those amino acids that have formed quite strong complexes with PS in the process of ethanol dehydration and leaves water-soluble and alcohol-soluble AA in the solution. It is worth noting that amino acids were not detected in the original PS samples. This fact is indicative of a high degree of PS purification from possible protein contaminants. The results of the experiment for the coprecipitation of PS with 20 amino acids are shown in Table 3.
The presented data demonstrate that all AA used in the experiment interacted with PS at different levels of efficiency. The fact that the process of bond formation de-

Table 3. Composition of amino acids detectable in the ethanol precipitate of PS (mcg/mg).
pends largely on the AA radical is beyond question. It has been established that homoglycans of acidic sugars (PU) bind AA more efficiently than homoglycans of neutral sugars (amylose) (124.46 and 32.20 mcg/mg PS, respectively). The experimental results are in good agreement with the quantum-chemical calculations (Table 2), which showed that the bond energy of the complexes AA radical end group—acidic saccharide carboxyl group is significantly higher (on average −5.00 kcal/mol) than the bond energy of the complexes AA-neutral saccharide hydroxymethyl group (on average −2.27 kcal/ mol). This, apparently, accounted for a more efficient formation of the AA-PU complexes. The adsorption rate of AA from the first class was up to 2.6 mcg/mg PU, whereas 7 AA from the fourth class showed the maximum PU affinity (on average 9.28 mcg/mg), which also confirms the results of the quantum-chemical calculations.
Acidic amino acids (Asp, Thr, Ser, Glu, Pro, Ala) bind more effectively to amylose (about 33% to AM vs. 23% to PU). In contrast, basic amino acids (His, Lys, Arg) were detected more often in the complexes with PU (about 30% with PU vs. 17% with Am). Neutral AA form complexes with PU in equal proportion (about 50%).
The results of the experiment for the abiogenic formation of peptide bonds between AA in the presence of PS are shown in Figures 2 and 3.
The presented data (Figures 2) testify that in the control (amino acids without PS), temperatures within the range 40˚C to 130˚C promote peptide bond formation in the process of AA dehydration. Maximum peptide bond formation was detected at temperatures over 100˚C (the Fox thermal proteinoids). The formation of peptide bonds was not detected in the control sample at 20˚C. However, temperature increase over 120˚C leads to the decrease in peptide bond formation. The presence of PS, especially acidic sugar homopolymers (PU), dramatically improved the effectiveness of this process (approximately by an order of magnitude) in the physiologically acceptable temperature range (20˚C - 60˚C), which indicates a significant catalytic activity of PS in terms of peptide bond formation. Gel-filtration (Figure 3) con-

Figure 2. Dynamics of peptide bond formation by means of the biuret reaction in the mixture of 20 amino acids under various temperature regimes, in the absence of PS (control) and in the presence of PS (PU, Am).
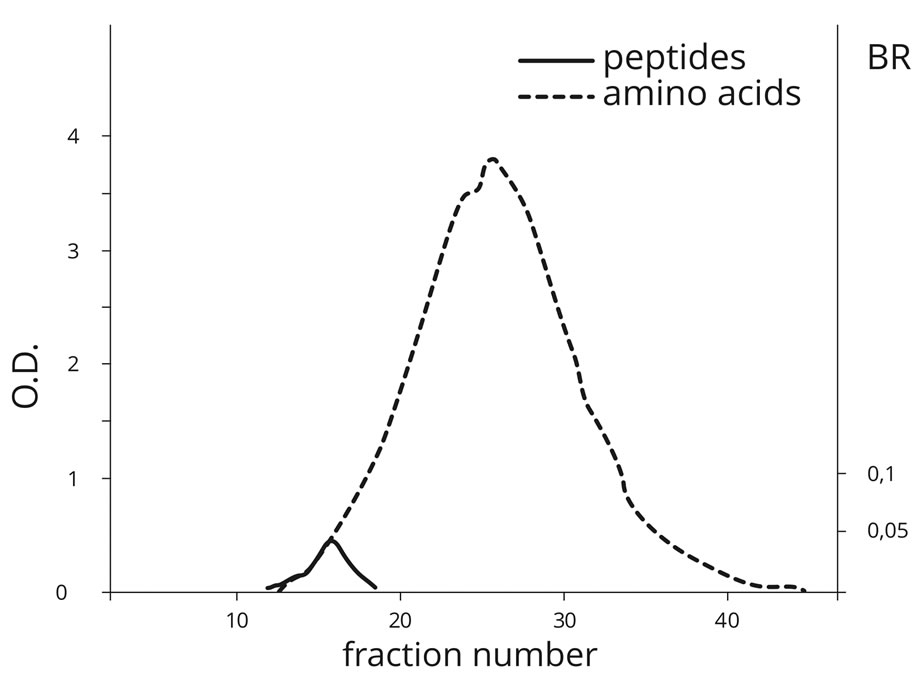
Figure 3. Gel filtration of the products obtained by temperature treatment (60˚C) on AA mixture in the presence of PS (PU) on Sephadex G-25. (Peptides detection is based on OD of the biuret reaction (BR) and AA detection is based on OD at 280 nm.)
firmed the fact of amino acid polymerization with the formation of peptide bonds. The peak of AA elution was detected in fractions 25 - 28, whereas the maximum of peptide bond was detected as early as in fraction 15. This attests to the formation of polypeptides, which are dozens of AA monomers in length. Peptide bonds were practically not formed in the absence of PS at temperatures below 40˚C.
3.2. NA Polymerization on PS
It was found that both acidic and neutral polysaccharides are capable of forming hydrogen bonds with nucleic acid bases. Table 4 demonstrates energy estimates of hydrogen bonds between the carboxyl group of polyuronides and the hydroxymethyl group of neutral saccharides with nucleotides obtained by the PM3 method. The computations show that the energy of hydrogen bonds between nucleotides and acidic polysaccharides is more than 3 times higher than the corresponding estimates for neutral polysaccharides, thus, theoretically predetermining their higher capacity for nucleotide adsorption. The calculations were confirmed experimentally (Table 5).
Indeed, homopolymers of acidic saccharides (PU) adsorb 3.5 times more nucleotides as compared to homoglycans NS (amylose).
As can be seen from the above, the specific adsorption of nucleotides on PS is beyond any doubt, according to the presented results and our earlier publications describing the physicochemical principles of these processes [11-14]. However, the question of nucleotide polymerization on a linear PS molecule has been undetermined. Our in silico study of mononucleotide interaction with a polysaccharide molecule showed that nucleotide monomers on PS are ideally oriented to form covalent bonds between them [12]. This should significantly stimulate
Table 4. Energy of hydrogen bonds between the carboxyl (PU) and hydroxymethyl groups (NS) of saccharides in a complex with nucleotides (PM3 method, kcal/mol).

Table 5. Quantity of deoxynucleotides detectable in the ethanol precipitation of oligosaccharides (mcg NA/mg PS).
the process of polynucleotide formation from monomers bound with PS. This assumption was verified in the series of experiments for the dehydration of PU complexes with AMP as it was described in the experimental section. These experiments were aimed at obtaining poly(A) oligomers with their further isolation using the specific oligo(dT) cellulose sorbent.
The control of our experiment was the approach used by L. Orgel for the synthesis of polynucleotides under the influence of thermal energy [5]. Polyuronides were used in the empirical sample in parallel with AMP. The results of the experiment are shown in Figure 4.
The results shown in Figure 4 demonstrate that AMP polymerization is practically undetected in the control sample dehydrated at a temperature below 100˚C, which corresponds to the data provided by L. Orgel. Indeed, the free AMP polymerizes at a temperature above 100˚C, while the nucleotide in the presence of PU shows the maximum NA polymerization rate in the temperature range 30˚C to 80˚C. Further temperature increase in the empirical sample resulted in the destruction of the polynucleotide.
As can be seen from the above, PU demonstrated a significant catalytic activity in the process of nucleotide polymerization within the physiological temperature range (20˚C - 60˚C), and apparently acted as the template for the arrangement of NA polymer molecules. It seems fair to assume that, that PU do not only adsorb nucleotides, but also facilitate the process of their polymerization when dehydrated. Abiogenic polymerization of nucleotides is about 10 times more intensive in the presence

Figure 4. The dynamics of poly(A) NA formation (chromategraphy on the oligo(dT) cellulose column) at different temperatures in the absence (control) and presence of PU (OD at 260 nm). Note: PU dehydration was carried out only in the presence of AMP.
of PU. NA concentration is increased in the PU-NA complexes in the subsequent processes of solution and repeated dehydration of the complexes with the mixture of nucleotides, as it is demonstrated in Table 6.
Our next objective was to isolate NA fragments formed on the PU by chromatography for their further analysis using the PCR method. As it is shown in Figure 5(A), the results of PU-NA complex chromatography on Sephadex G-25 demonstrate that high-polymer PU components were eluted predominantly before fraction 15. Low-polymer components were eluted between fractions 16 and 22 in smaller quantities than the high-polymer ones. The monomers eluted after the 23th fraction predictably were not represented in the samples, since ethanol precipitation of PS-NA leaves nucleotide monomers in the solution.
As it is shown in Figure 5(B), the spectrophotometric analysis of the eluted complexes demonstrates that the low-polymer fraction is rich in nucleotides, as the OD ratio A260/A220 fluctuates within the range of 0.8 - 1.0 in these samples, whereas for the high-polymer fraction it is 0.5. These results suggest the presence of abiogenically formed DNA in the fractions containing low polymer fragments.
To verify the presence of “correct” DNA chains recognized by specific enzymes in the test sample and its characteristic, we carried out an analysis using PCR. The NA polymers of fractions 16 - 22 after the chromatography oh Sephadex G-25 were used in the experiment. The results of the experiment are shown in Figure 6.
As Figure 6 demonstrates, the primers themselves (Lanes 1) can serve as the template for the synthesis of high-polymer DNA. The reaction conditions facilitate the accumulation of the products at initially low template concentration due to the properties of the primers and to the presence of some DNA oligomers in the reaction mixture. Moreover, the introduction of an additional control—human DNA (Lane 2) to the experiment allows us to deny the presence of human DNA in the tested samples and to observe low ability of the primers to “self-prime” in the presence of the given DNA.
The acceleration of the polymerase activity is observed in the test sample (Lane 3) in the presence of AFK-3 primer. The new product, which is a few hundreds of nucleotide pairs in length, cannot be explained by human DNA contamination (most likely in the PCR process). Since the DNA product in the tested sample is formed in the presence of only one of the primers, it can be assumed that the amplicon, and therefore, the original DNA have low “complexity”, i.e. have limited number of primary structure variants. The latter, as is well-known, is typical for DNA tandem repeats.

Table 6. Optical characteristics of PS-NA complexes subjected to dehydration once (I), twice (II), three times (III) (260/220 nm).
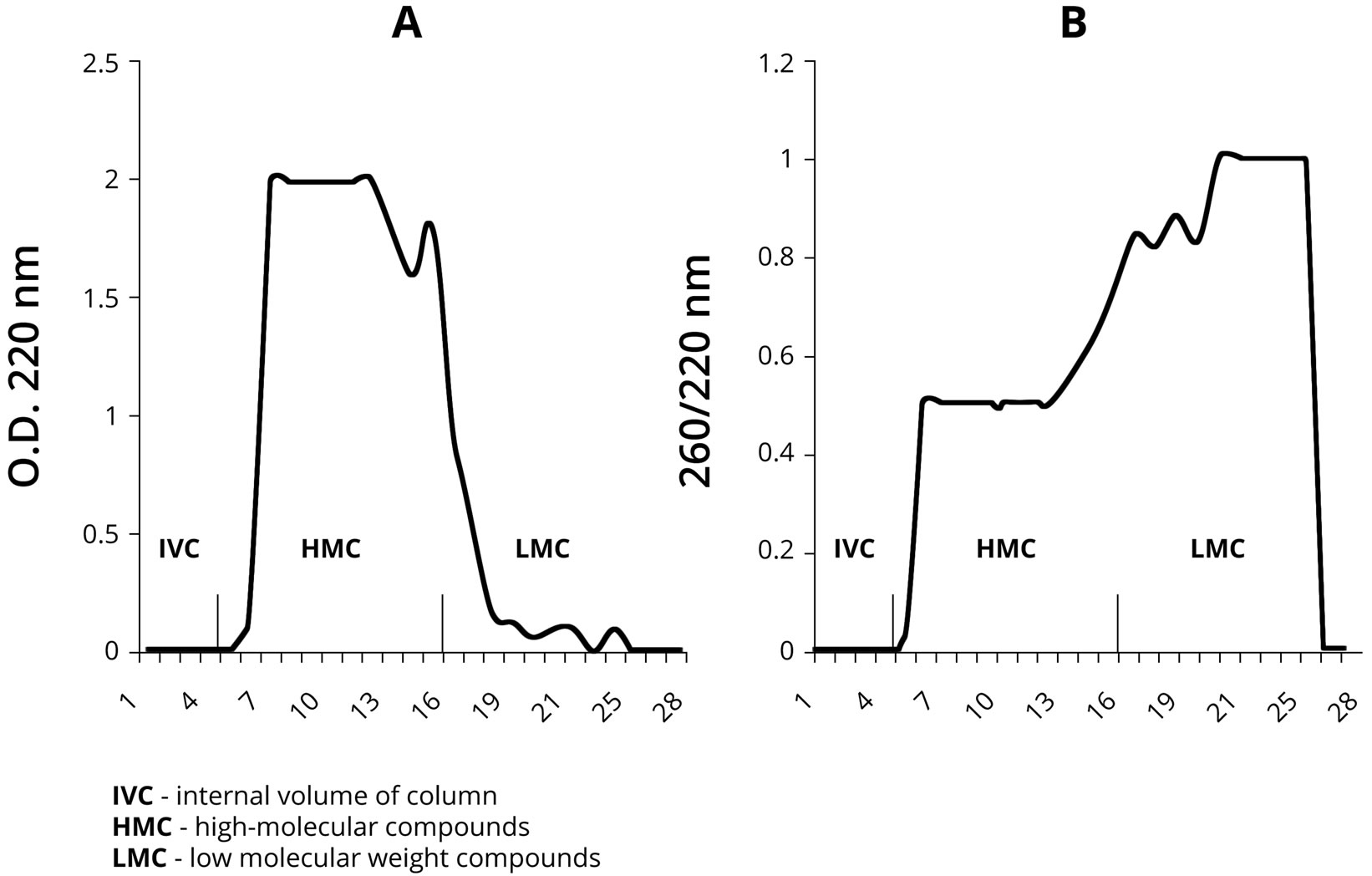
Figure 5. PU-NA complex chromatography on Sephadex G-25. A—represents detection based on OD at 220 nm, and B—represents detection based on OD 260/220 nm measurements.

Figure 6. 1. The results of DNA-RAPD with the DNA preparation synthesized on PU. Control (primer); 2. Human DNA + primer; 3. DNA synthesized on PU, + primer.
The results of PCR with AFR-3 primer on PU and on the isolated NA fragments synthesized on PU are shown in Figure 7.
The Figure 7 demonstrates that the primer itself (Lane 1) can serve as the template for high-polymer DNA synthesis as per the above procedure, promoting the product accumulation at initially low concentration of the template due to the properties of the primer itself and the presence of the admixture of DNA fragments of an unidentified origin.
The activity of the Taq-polymerase used in the PCR reaction was not detected in the samples containing PU and nucleotide (Lane 3) as well as in the pure PU (Lane 2). It may suggest the absence of NA fragments in the original PU samples and the ability of PU in high concentrations to suppress enzyme activity. Alternatively, the PU from fractions 16 - 22 (Figure 5(A)) stimulated enzyme activity. This fact attests the presence of DNA molecules in the given polysaccharide fractions (Lane 4). The synthesized product, which is a few hundred nucleotide pairs in length, does not result from human DNA contamination, as was mentioned above (Figure 6). No visible quantities of the product are detectable in Lane 5 demonstrating the PCR result in the conditions of selfpriming of isolated NA synthesized on PU. It can be caused by significant variations throughout the length of the analyzed NA sequences formed on PU.
As can be seen from the above, the PCR method confirmed the presence of DNA oligomers of varying sizes synthesized on PU. According to preliminary data they have a “simple”, most probably tandem structure. We found that that the given NA sequences do not relate to human DNA. The nature of the given NA calls for further research [14].
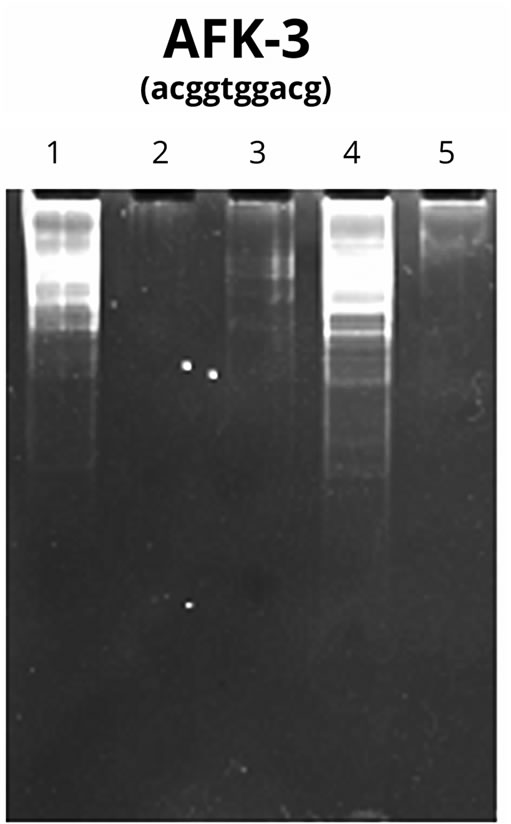
Figure 7. DNA-RAPD results with the preparations of NA synthesized on PU Lanes: 1—control (AFK-3 primer); 2— AFK-3 primer + PU; 3—AFK-3 primer + PU-nucleotide mixture; 4—AFK-3 primer + oligo nucleotides from PU-NA complex after dehydration; 5—isolated oligonucleotides from PU-NA complex in the absence of the primer.
3.3. NA Aminoacylation in the Process of AA and NA Interaction with PS
Since PS can simultaneously bind both nucleotides and amino acids, there is a need to examine possible interactions of AA with NA that are in complex with PS. When hydrogen bonds are formed between NA and PS bases, a complex is created at a distance of 5.7 - 7.5 Å from the zone of hydrogen bond formation between NA and PS to the carbon at the 2nd, 3rd and 5th position of the saccharide in nucleotide A.
Apart from the charge, another significant characteristic of AA radicals is the size. All proteinogenic amino acids can be subdivided into three groups: those with a “long” radical, 6.7 - 7.4 Å (Arg, Phe, Lys, Tyr), a “medium” one, 4.0 - 5.9 Å (Trp, Gln, Glu, Met, Asn, Asp, His, Leu, Ile, Val), and a “short” one, 2.1 - 3.8 Å, (Ser, Thr, Pro, Cys, Ala, Gly). The size, as well as the charge, can influence bond formation process. If the end group of AA radical forms hydrogen bonds with the PS monosaccharide, then the aminocarboxymethyl group of the amino acid can be located in the zone of the OH group, at the 2’, 3’ carbon of nucleotide A ribose (Figure 8) or phosphate, at the 5’ carbon.
The carboxyl group of AA located in this way in the PS-NA-AA complex enables the aminoacylation process at the above mentioned hydroxyls or phosphate of NA saccharide carbon ring. This assumption was verified by the quantum-chemical calculations (Table 7), according to which the formation of the AA-NA complex becomes possible when interacting with OH groups of the 2’ and 3’ carbons of NA ribose, in complex with PS. A fragment of RNA CCA in complex with AA and tetrasaccharide NS-NS-AS-AS was used in the calculations.
As can be seem from the above (Table 7), abiogenic aminoacylation of the terminal A nucleotide ribose is hypothetically possible in terms of the length of the formed
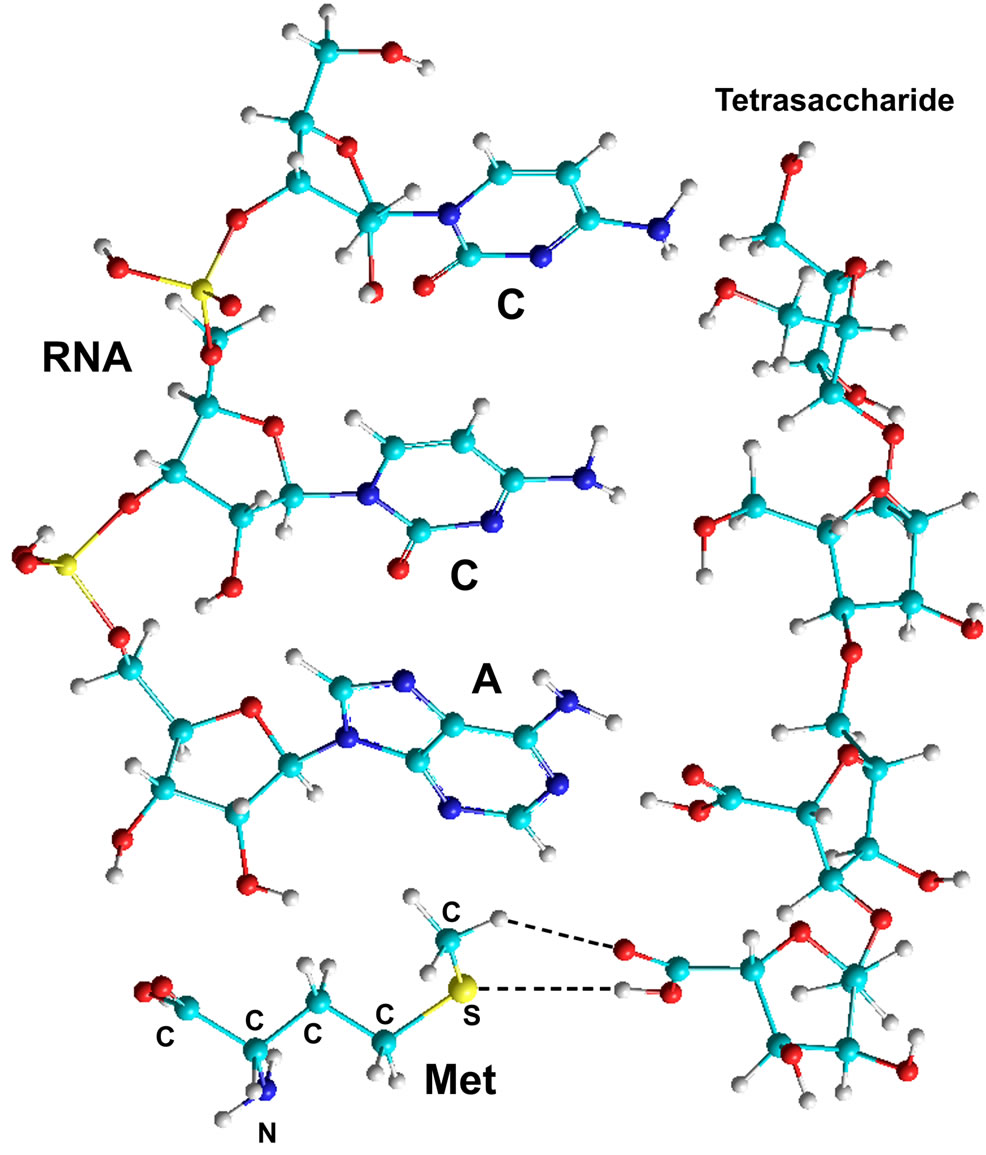
Figure 8. Quantum-chemical model of the complex RNA (CCA)- Methionine-Tetrasaccharide (NS-NS-AS-AS) (calculations by the PM3 method).
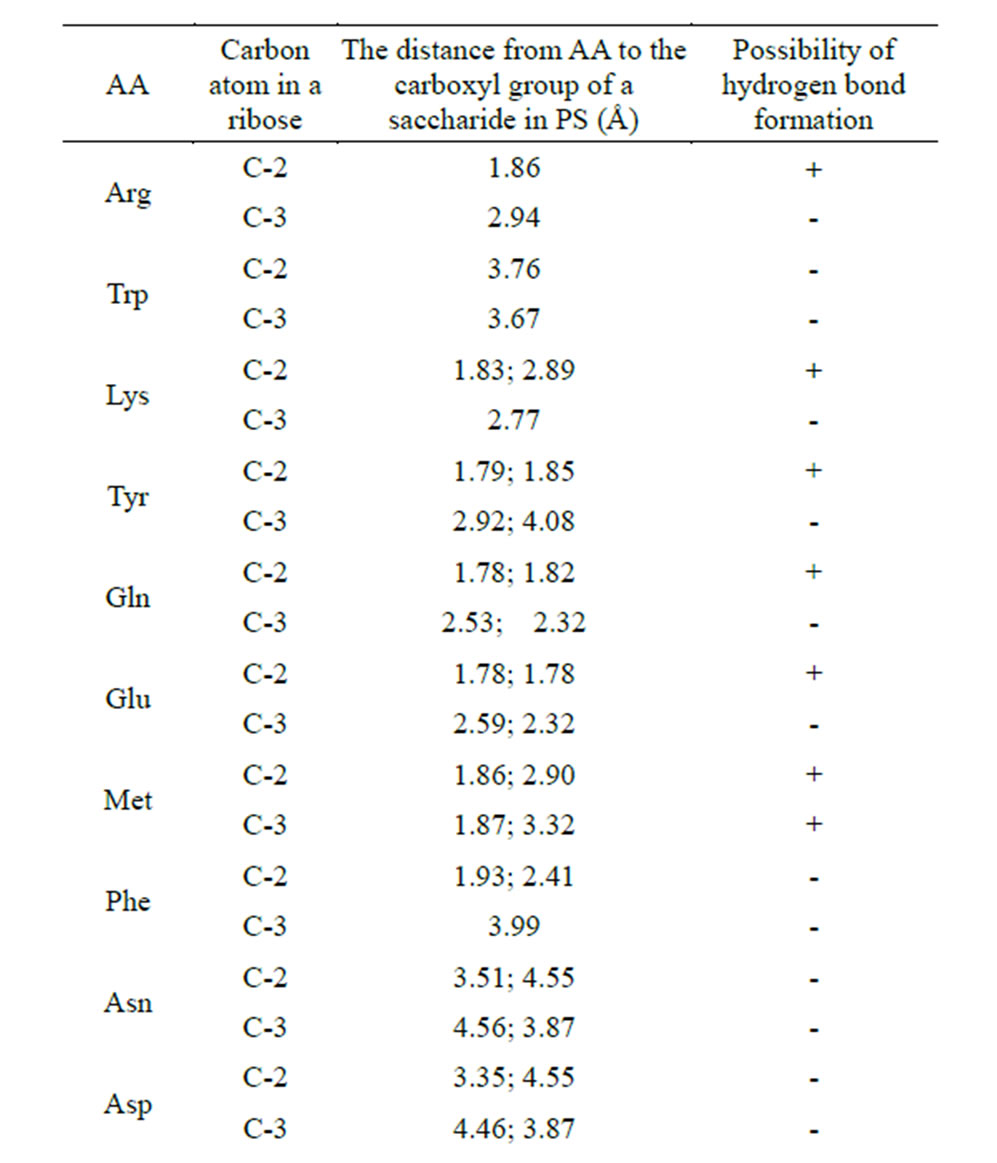

Table 7. Calculation of the distances between the interacting atoms of the AA radical end group and the saccharide carboxyl group in the complex NA-AA-PS (Å)(AA is covalently bound to the hydroxyl of NA ribose at the С2 and С3.
hydrogen bonds (the bond length is less than or equal to 1.87 Å) for six amino acids—Arg, Lys, Tyr, Gln, Glu, Met. Furthermore, Met and Gln are able to carry out this process at the hydroxyls 2 and 3 of the ribose. Energy indices of the hydrogen bonds formed between the end group of AA radical and the carboxyl group of the saccharide also confirm the possibility of this process, since it goes with heat release (Table 8).
It is worth noting that there is a possibility of aminoacylation for Tyr at С2 hydroxyl of the ribose, whereas there is no such possibility for Trp. Since these two AA can be easily detected in the complexes by the spectral analysis, we conducted additional calculations to test the ability of the given AA to aminoacylate the phosphate at the С5-position of the ribose.
The results of the calculations are given in Table 9 (Figures 9 and 10), which shows that theoretically Tyr is able to form bonds with the phosphate at C5. In contrast, Trp has no such possibility in the AMP variant. However, in the ADP and ATP variants Trp amynoacylation is quite possible.
In order to prove empirically the possibility of abiogenic aminoacylation in accordance with the performed computations, dehydration of the PS—poly-A RNA-AA complex was conducted. The results of the experiment as exemplified by two AA are shown in Table 10. The polyA RNA fragments (Section 3.2) obtained abiogenically were re-dehydrated in the presence of PU and amino acids (Tyr or Trp) at a temperature of 60˚C. Then the

Table 8. Total energy of hydrogen bonds, which coordinate AA as a separate molecule when the end group of the AA radical interacts with the carboxyl group of the saccharide in the process of the NA-AA-PS complex formation.
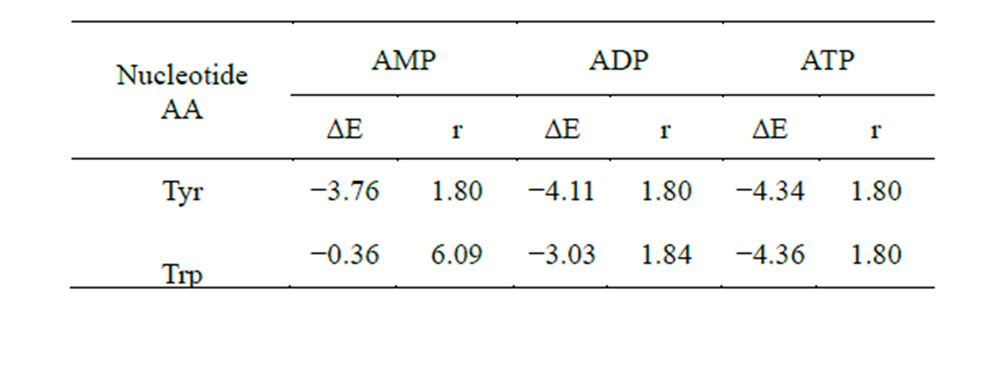
Table 9. Energy and length of H-bond hydrogen (AA)—oxygen (phosphate at С5 of the ribose) in the system AA-nucleotide-disaccharide (ΔЕ, kcal/mol and r, Å).
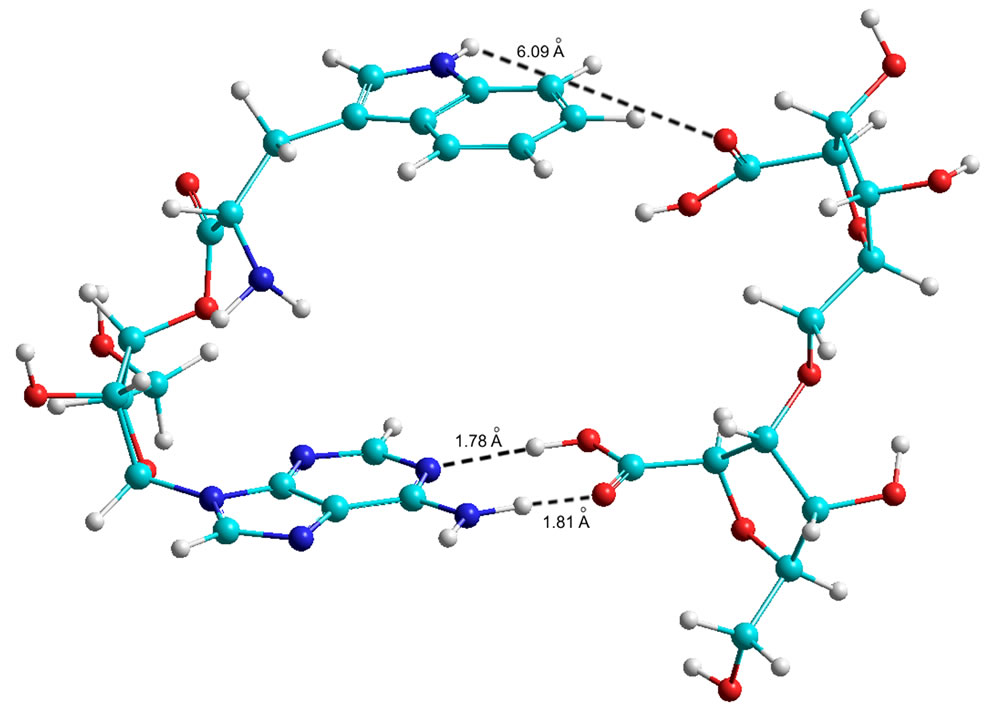
Figure 9. Quantum-chemical model of the 5’ AMP-Trp-Disaccharide complex (AA at С5 of the ribose).

Figure 10. Quantum-chemical model of the 5’ AMP-Tyr-Disaccharide complex (AA at С5 of the ribose).
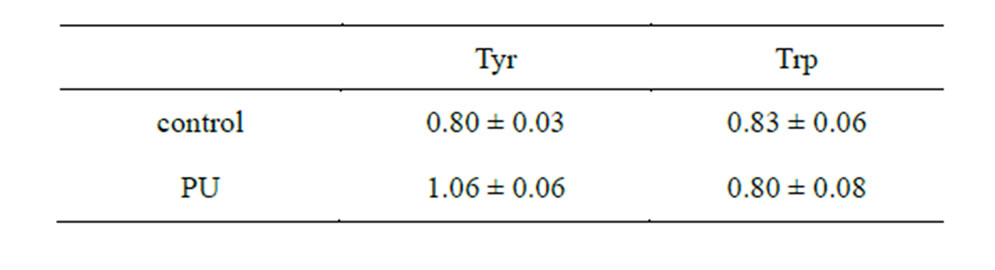
Table 10. Characteristic of abiogenically obtained poly-A RNAAA complexes under dehydration at a temperature of 60˚C (OD ratio 280/260 nm).
NA-AA complexes were isolated by an affinity chromatography on oligo (dT) cellulose columns by the poly-A fragment.
Samples of poly-A RNA of the corresponding AA dehydrated in the absence of PU served as the control. The presence of AA in the structure of RNA polymers was detected spectrophotometrically (Table 10). In contrast to AMP (260 nm), the above mentioned AA have the maximum absorbance at 280 nm (Figure 11).
As can be seen from the table above, a significant increase in absorbance at the wavelength of 280 nm is observed only in the oligo-A sample, subjected to dehydration in the presence of PU and tyrosine (OD ratio 280/ 260 equals to 1.06). This suggests the presence of AA in
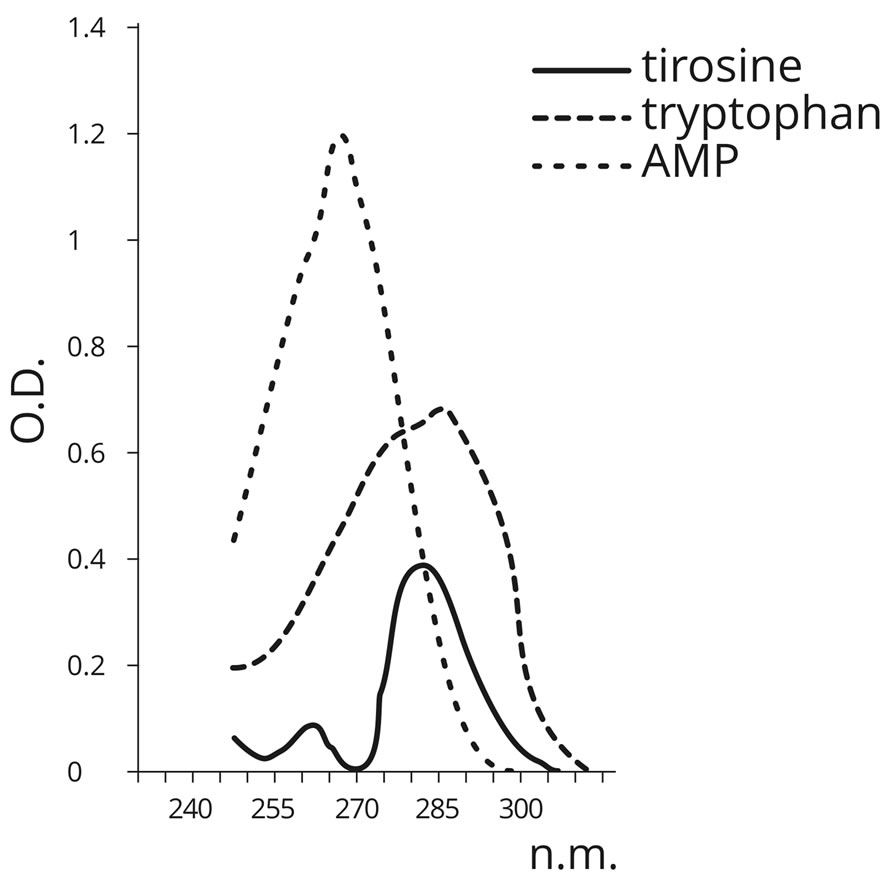
Figure 11. Absorbance spectra of AMP, tyrosine and tryptophan.
the RNA. Tryptophan did not form a complex with oligoA in the presence of PU. These data verify the conclusions about the abiogenic aminoacylation of nucleotide A ribose based on the quantum-chemical calculations. In the experiment outlined above, nucleotide A retained its ability to bind to nucleotide T of the sorbent (oligo (dT) cellulose), which suggests that the base does not participate in forming bonds with AA. Consequently, AA detected in the RNA complexes is connected to the ribose of the nucleotide at С2 or С5. Therefore, quantum-chemical calculations attest that aminoacylation of nucleoside monophosphates by tyrosine is possible both at the С2 and С5 positions. Tryptophan is not able to aminoacylate nucleoside monophosphates either at the С2 or С5 positions. It is most likely that the AA determined in the tyrosine-oligonucleotide complexes utilized the opportunity to aminoacylate the nucleotide at the С2 or С5 positions in the ribose, which is supported by the experimental results. It should be noted that PS aminoacylate phosphoric acid residue more efficiently at the С5 position of nucleoside triphosphates.
As can be seen from the above, experimental and calculation data demonstrate that there are quite strict patterns of PS interaction with AA and NA. PS are not only able to conduct adsorption and polymerization of the above noted monomers of biopolymers, but also, under certain conditions, to perform abiogenic aminoacylation of a nucleotide ribose both at the C2-C3 and C5 positions.
4. DISCUSSION
Regardless of the opinion diversity, it is highly plausible that sugars, amino acids, phosphorous and nitrogen compounds (NA bases) existed at the beginning of the molecular evolution. Sugar oligomers are formed in an auto-catalytic manner, abiogenically, in aqueous solutions, since they are synthesized from formaldehyde and polymerized without dehydration [2]. Amino acid and NA polymerization, in its turn, proved to be rather simple under the conditions of dehydration, under the influence of thermal energy, in the presence of PS, in the physiologically acceptable temperature range, as it was demonstrated in the present work. S. W. Fox [4] had previously established the possibility in principle of AA polymerization when their crystal mixture is exposed to temperatures above 100˚C. Abiogenic formation of thermal nucleotides at temperatures above 100˚C was also suggested by L. Orgel [5]. Due to such high temperatures of biopolymer formation, these abiogenic processes were restricted only to volcanic activity zones, where temperature conditions did not contribute to the stabilization of biogenic systems. Moreover, the excess of thermal energy could lead to polymer destruction. For the origin of primary living systems, similar to coacervates, a catalyst was needed to promote the polymerization process in the temperature range that would not destroy the forming polymers. It was oligosaccharides which, due to their abundance in the initial conditions at the beginning of the molecular evolution of biopolymers [2], could initiate the above-mentioned processes, which, in their turn, enabled the described reactions to prevail at the initial stages of the Earth atmosphere formation. It is hardly probable that clays and minerals could perform this function. Since minerals are characterized by a significantly lower adsorption activity as compared to PS, they are not capable of evolving in terms of their structure and do not show a catalytic activity in AA and NA polymerization. As we have demonstrated in this work, PS do not have such disadvantages.
Since polysaccharides adsorbed both AA and nucleotides (the latter demonstrated selectivity in bond formation similar to NA complementarity), it was PS that could act as the primary information system, which helped to combine these two groups into one, thus, initiating the abiogenic aminoacylation of nucleotides. According to the results of the quantum-chemical calculations and empirical research, it was established that for some amino acids this process could be carried out abiogenically, not involving enzyme systems, at 2-3’ end of the ribose in the nucleotides. Aminoacylation with the help of PS is possible practically for all biogenic amino acids at the 5’ end of nucleoside triphosphates. Minispiral forms of RNA (the precursors of the modern tRNA [21]), whose terminal nucleotide is attached to the amino acid, were formed on PS on the principle of PS-NA complementaryty. This process could be a starting point for the template assembly of primary polypeptides abiogenically on oligosaccharide and nucleotide templates. It was PS that were able to function as the catalyst that promoted the formation of both NA and AA polymers and the modern triplet genetic code. Therefore, at the beginning of molecular evolution PS were responsible for combining nucleotides and amino acids into a single system of genetic information formation via three types of biopolymers—NA, protein and PS.
Further research in this direction may bring us closer to the understanding of the mechanisms of genetic code formation in the course of molecular evolution of biopolymers and, furthermore, to evaluate the importance of oligoand polysaccharides in modern biosystems, since in vivo synthesis of at least some of them are templatebased. The obtained data may help answer one of the principal questions about life origin on the Earth—in what way, how and when NA combined with polypeptides to form the modern system of accumulation, storage, transfer and implementation of genetic information.
ACKNOWLEDGEMENTS
This work was supported by the “Plazan” Closed Joint-Stock Company, Moscow.
REFERENCES
- Miller, S.L. and Urey, H.C. (1955) Production of some organic compounds under possible primitive Earth conditions. Journal of the American Chemical Society, 77, 2351. http://dx.doi.org/10.1021/ja01614a001
- Parmon, V.N. (2002) Prebiotic phase of life origin. Vestnik Rossiskoi Akademii Nauk, 72, 976-983.
- Rezanov, I.А. (1985) Great calamities in the Earth history. Mir, Мoscow.
- Fox, S.W. and Dose, K. (1972) Molecular evolution and the origin of life. S.A. Freeman Co., San Francisco.
- Orgel, L.E. (1998) The origin of life—A review of facts and speculations. Trends in Biochemical Sciences, 23, 491-495. http://dx.doi.org/10.1016/S0968-0004(98)01300-0
- Oparin, А.I. (1968) Life, its nature, origin and development. Nauka, Moscow.
- Bernal, J. (1969) The origin of life. Мir, Moscow.
- Zimina, N.P., Rykova, V.I. and Arkhipov, I.A. (1992) Animal tissue proteoglycans as irregular biopolymers: Informativeness of structure and control of biosynthesis. Uspehi Sovremennoi Biologii, 112, 571-588.
- Silbert, J.E. (1982) Structure and metabolism of proteoglycans and glycosaminoglycans. Journal of Investigative Dermatology, 79, 31-37. http://dx.doi.org/10.1038/jid.1982.7
- Sugahara, K. and Kitagawa, H. (2000) Recent advances in the study of the biosynthesis and functions of sulfated glycosaminoglycans. Current Opinion in Structural Biology, 10, 518-527. http://dx.doi.org/10.1016/S0959-440X(00)00125-1
- Zimnitskii, A.N., Bashkatov, S.A. and Urazbaev, V.N. (2005) DNA tandem repeats and the concept of template synthesis of proteoglycans. Labirint, Мoscow.
- Zimnitskii, A.N., Bashkatov, S.A. and Urazbaev, V.N. (2007) Interaction between nucleic acids and glycans: In silico and in vitro. Biophysics, 52, 282-287. http://nanoderm.ru/img/userimg/statyi/magazine_biofizika_eng.pdf http://dx.doi.org/10.1134/S0006350907030049
- Zimnitskii, A.N. (2012) Concept of template synthesis of proteoglycans. In: Dr. D. N. Karunaratne, Ed., The Complex World of Polysaccharides. http://www.intechopen.com/books/the-complex-world-of-polysaccharides/concept-of-template-synthesis-of-proteoglycans/ http://dx.doi.org/10.5772/48085
- Zimnitskii, A.N., Bashkatov, S.A., Chemeris, A.V., Yamidanov, R.S. and Urazbaev V.N. (2014) Polysaccharides as initiators of nucleic acid polymerization. American Journal of Molecular Biology, 4, 20-25. http://dx.doi.org/10.4236/ajmb.2014.41004
- Sterwart, J.J.P. (1989) Optimization of parameters for semiempirical methods. II. Applications. Journal of Computational Chemistry, 10, 221-261. http://dx.doi.org/10.1002/jcc.540100209
- Schmidt, M.W., Baldridge, K.K., Boatz, J.A., Elbert, S.T., Gordon, M.S., Jensen, J.H., Koseki, S., Matsunaga, N., Nguyen, K.A., Su, S.J., Windus, T.L., Dupuis, M. and Montgomery, J.A. (1993) General atomic and molecular electronic structure system. Journal of Computational Chemistry, 14, 1347-1363. http://dx.doi.org/10.1002/jcc.540141112
- Orekhovich, V.N. (1968). Modern methods in biochemistry. Meditsina, Moscow.
- Osterman, L.А. (1981) Protein and nucleic acid research methods. Electrophoresis and ultracentrifugation. Nauka, Мoscow.
- Gilyazetdinov, S.Y. and Zimnitskii, A.N. (1986) Composition of poly(А+)-RNA population in the roots of corn heterotic hybrids and their original strains. Physiology of Plants, 33, 769-777.
- Singer, M. and Berg, P. (1998) Genes and genomes. Mir, Moscow.
- Moras, D. (1993) Structural aspects and evolutionary implications of the recognition between tRNA and aminoacyl-tRNA-synthetases. Biochemie, 75, 651-657. http://dx.doi.org/10.1016/0300-9084(93)90095-A