Natural Science
Vol.4 No.12A(2012), Article ID:26150,4 pages DOI:10.4236/ns.2012.412A141
Prot-Class: A bioinformatics tool for protein classification based on amino acid signatures
![]()
1School of Electrical Engineering and Computer Science, Russ College of Engineering, Ohio University, Athens, USA; *Corresponding Author: lichtenj@ohio.edu
2Department of Environmental and Plant Biology, College of Arts and Sciences, Ohio University, Athens, USA
3Biomedical Engineering Program, Ohio University, Athens, USA
4Molecular and Cellular Biology Program, Ohio University, Athens, USA
Received 22 June 2012; revised 25 July 2012; accepted 12 August 2012
Keywords: Bioinformatics; Protein Classification; Arabidopsis
ABSTRACT
Knowledge about characteristics shared across known members of a protein family enables their identification within the complete set of proteins in an organism. Shared features are usually expressed through motifs, which can incorporate specific patterns and even amino acid (AA) biases. Based on a set of classification patterns and biases it can be determined which additional proteins may belong to a specific family and share its functionality. A bioinformatics tool (Prot-Class) was implemented to examine protein sequences and characterize them based upon user-defined AA composition percentages and user defined AA patterns. In addition the tool allows for the identification of repeated AA patterns, biased AA compositions within windows of user-defined length, and the characteristics of putative signal peptides and glycosylphosphatidylinositol (GPI) lipid anchors. ProtClass is general purpose and can be applied to analyze protein sequences from any organism. The Prot-Class source code is available through the GNU General Public License v3 and can be accessed via the Google Code Repository: http://code.google.com/p/prot-class.
1. INTRODUCTION
The genomics era has generated enormous amounts of nucleotide and amino acid sequence data, resulting in the identification of gene sequences and their corresponding protein sequences. The identification and characterization of genes and their associated protein functions remain a major goal in biology. Associated with this goal is the identification and classification of genes/proteins into related families and subfamilies. This paper presents a bioinformatics tool (Prot-Class), which analyzes protein sequences and classifies them using user-defined amino acid (AA) signatures, given through AA composition percentages and AA query sequences.
Prot-Class is useful in identifying proteins with known signatures, but can also be useful in the characterization of novel proteins. Once such analysis is completed, the protein sequences can be analyzed further by Prot-Class to reveal repeated AA sequences, biased AA compositions within a user-defined window, and putative signal peptide sequences responsible for extracellular secretion and glycosylphosphatidylinositol (GPI) lipid anchor addition sequences allowing for attachment of the protein to the plasma membrane. Prot-Class has advantages over conventional BLAST searches [1] used for identifying related gene/protein families and subfamilies, particularly for proteins with AA compositional biases or characteristic sequence motifs, as Prot-Class allows for the identification of related genes/proteins showing low sequence similarities.
Prot-Class was originally developed to mine the 28,952 protein sequences generated from the Arabidopsis thaliana genome project for hydroxyproline-rich glycolproteins (HRGP) sequences. HRGPs are a family of extracellular matrix proteins found throughout the plant kingdom which function in various aspects of plant growth and development. The HRGP family is divided into three families, the arabinogalactan-proteins (AGPs), extensins (EXTs), and proline-rich proteins (PRPs). The tool was used (under the name Bio Ohio) to successfully mine the data, and the biological implications of this work are published [2]. This paper describes how the tool works and illustrates its biological utility as a generalpurpose application.
There are two general approaches to classifying proteins: Alignment-based and non-alignment based. Nonalignment based methods, such as the one described in this paper, also include using string kernels and profiling distributions of four contiguous amino acids in known protein families (e.g. a ≥50% bias of proline, alanine, serine and threonine in AGPs). Alignment based methods include building blocks of alignments and matching unknown protein sequences against these blocks to correlate many connections at once.
The method of protein classification by string kernels has the benefits of being computationally cheap and does not require prior knowledge of the sequences. Classifying in this manner has produced highly accurate results, but not substantially better than any other methods [3,4]. Another non-alignment based method is to profile a protein family using its distribution of four amino acids, then profile an unknown sequence, and finally match the profile signatures [5]. For alignment-based methods, there is generally the concern that it is computationally expensive and not scalable in regard additional sequences being submitted to the various databases. In order to address this concern, a method of combining highly conserved locally aligned sequences into blocks has been developed. This allows for the unknown sequence to be matched against these blocks to facilitate faster alignment searching [6,7]. None of these alignment-based methods are done in a modular manner that allows for additional plug-in of functionality. The modular approach implemented by Prot-Class, however allows for additional functionality to be added easily. Prot-Class offers the benefit of allowing a user to add annotations, to save them for further work, and to download them. Prot-Class also integrates a set of important software features into one tool and allows for studying any family of proteins.
2. METHODS
Prot-Class, applied as a tool for the classification of proteins based on AA signatures (e.g., AA biases and/or string-based patterns), has nine major modules that communicate via an integrated framework (Figure 1).
The string-based pattern-matching feature of ProtClass allows the use of protein motifs, manifested as straightforward subsequences or short degenerate AA sequences using Perl-based regular expressions, as pattern-based classifiers in order to assign specific proteins with predefined labels. With respect to the EXT subfamily of the HRGPs in Arabidopsis, the motifs SP3 and SP4 (SPPP and SPPPP, respectively) were identified and used as EXT classifiers in Prot-Class [2].
Prot-Class allows the user to select or modify a spe-
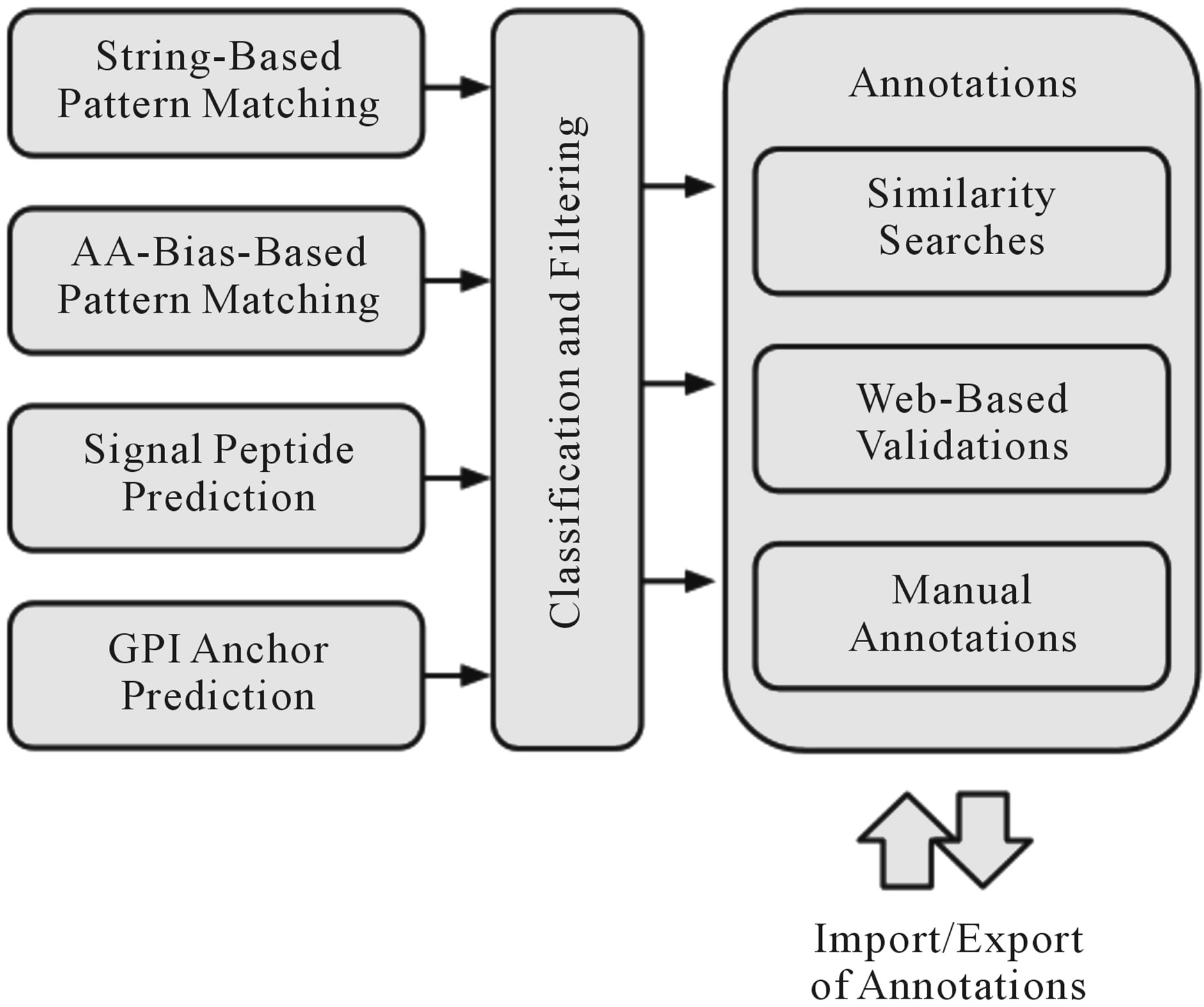
Figure 1. Workflow diagram of the Prot-Class framework.
cific AA bias. This AA bias-based pattern matching feature acts as a filter on the AA composition of a protein sequence. If a user-specified set of AAs covers at least a user-specified percentage of the sequence, the corresponding protein is classified as belonging to the family or subfamily in question. An application of the AA bias filter is presented in Schultz et al. [8], where a bias of ≥50% for proline (P), alanine (A), serine (S) and threonine (T) was required to classify a sequence as being a member of the AGP subfamily within the family of HRGPs.
The secretion of HRGPs to the plant cell wall is controlled by N-terminal signal peptides, which allow proteins to enter the cellular secretory pathway associated with the endoplasmic reticulum and Golgi apparatus. In addition, some AGPs are known to be associated with the plasma membrane through the presence of a C-terminal GPI lipid anchor. Signal peptide prediction can be achieved through existing approaches available through the opensource community. Prot-Class is using the BioPerl module Sigcleave. The GPI anchor prediction, native to the presented tool, is conducted by detecting and filtering possible omega sites (i.e., sites where the protein is cleaved and joined with the GPI lipid anchor) in a generally hydrophobic, C-terminal portion of a protein. Annotations for both results can be automatically corroborated by web-based validations using the SignalP tool [9,10] and the Plant big-PI predictor website [11] respectively.
In order to provide validations and annotations for predictions based on AA biases, string-based patterns, signal peptides and GPI anchors, Prot-Class conducts BLAST similarity searches for each specific protein that is predicted to be a member of the family. This automated step allows the identification of other potential family members that could not be detected due to threshold restrictions and also provides insight into the validity of the prediction through a comparison against the annotated functionality of the similarity matches. The impact of string-based patterns is represented through the breakdown of all repeated subsequences within a specific protein. In addition to the automatic annotations for the proteins, the user can manually annotate the classified sequences. The resulting predictions, classifications and annotations are displayed in Prot-Class via standard HTML tables. The annotated sequence information can be exported and shared between researchers and searched for specific keywords.
The Prot-Class software consists of an object oriented framework for dynamically configuring and running complex in-silico experiments that are well documented, repeatable, and easily extended. In order to support the complex classification, various parts of the framework are parallelized using the Perl Parallel: Fork Manager. The base class of the application is an operational manifest handling operations in regard to the motif classification tasks. These functions include the parsing of genomic databases and logging of results. All specific classes of experiments are derived from this “manifest” base class and implemented using specific logic rules.
The experimental framework is written in Perl and hosted to run as a web service. The source code is available through the Google Code Repository (http://code. google.com/p/prot-class/) under the GNU General Public License v3.
3. RESULTS
HRGP families contain unique biased AA compositions and/or AA sequences that make them ideally suited for identification with the Prot-Class program. AGPs are rich in P, A, S, and T. In order to identify putative AGPs from the Arabidopsis proteins, sequences with a PAST content of 50% or more were examined. Since some AGPs are known to be short, sequences having a PAST content of 35% or more and being 50 - 90 AAs in length, were also considered. The Arabidopsis protein sequences were also examined with respect to the presence of fasciclin sequences in order to identify fasciclin-like AGPs (FLAs). In addition to the discovery of AGPs, Arabidopsis proteins were classified with respect to other HRGP related subfamilies (EXTs and PRPs).
All HRGP sequences were subsequently examined for repeating AA sequences, signal peptides, and GPI anchor addition sequences. The resulting HRGPs identified by the program are shown in Table 1. Table 1 also shows HRGPs identified by previous studies [8,12].
This tool resulted in the identification of several new AGP, EXT, and PRP family members as well as confirmed the existence of several of these sequences from
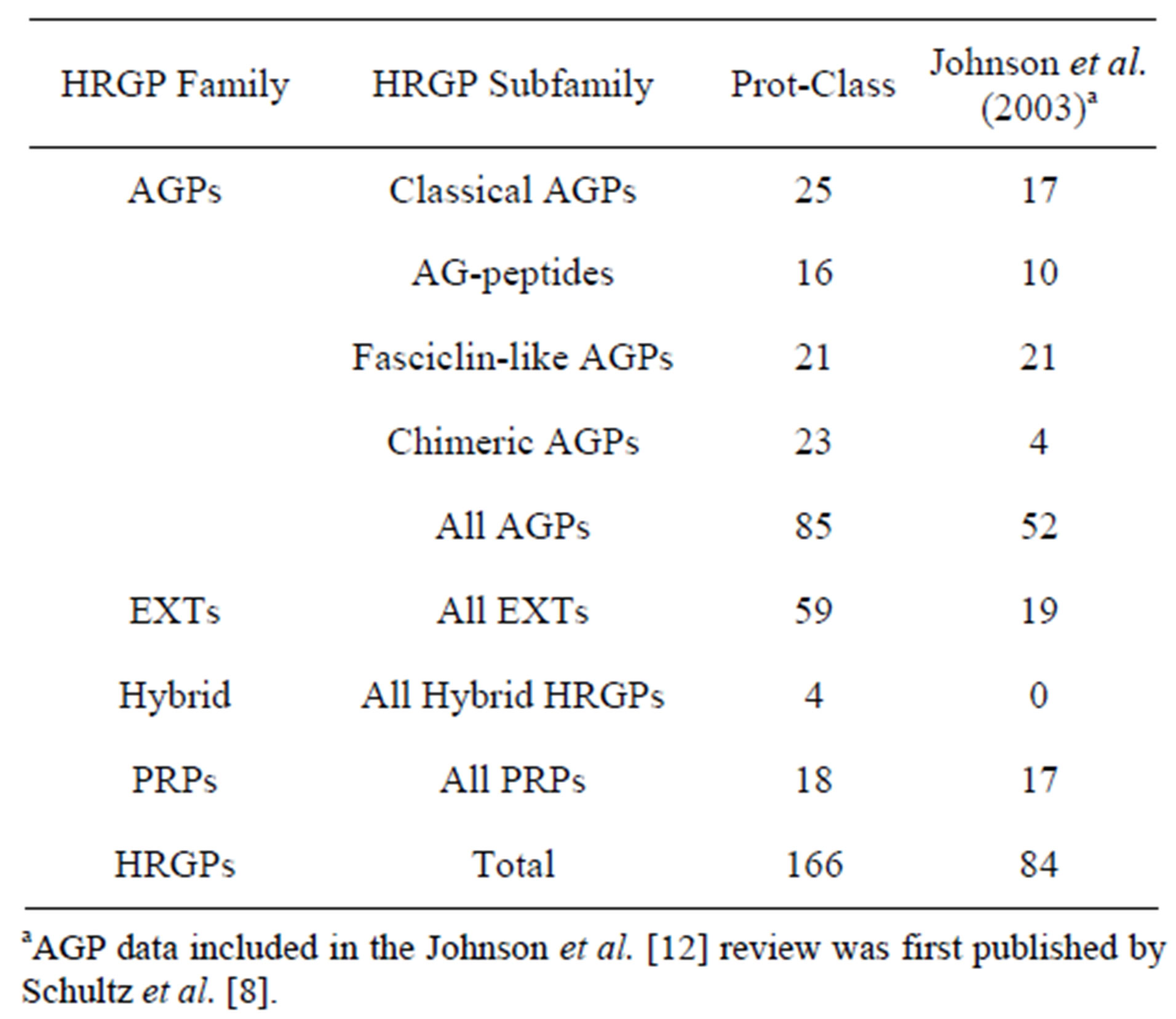
Table 1. Comparison of HRGPs identified by Prot-Class to HRGPs identified in previous studies.
previous studies. Schultz et al. [8] previously utilized a bioinformatics approach to identify candidate AGP genes from Arabidopsis. In contrast to this study, only 52 AGPs [including 17 classical AGPs, 10 AG-peptides, 21 (chimeric) FLAs and four other chimeric AGPs] were identified.
In conclusion, this tool was effective in confirming and identifying new HRGP gene/protein family members in Arabidopsis. Use of this tool to identify HRGPs in other plant species is now possible. This tool, however, is of general use and can be easily applied by the user to identify other protein family members in any species having protein sequence data.
4. ACKNOWLEDGEMENTS
Funding: The authors acknowledge the financial support of the Stocker Endowment, Ohio University’s Graduate Research and Education Board (GERB), the National Science Foundation (grant no. 0918661) as well as the Choose Ohio First Initiative of the University System of Ohio.
REFERENCES
- Altschul, S.F., Gish, W., Miller, W., Myers, E.W. and Lipman, D.J. (1990) Basic local alignment search tool. Journal of Moecular Biology, 215, 403-410.
- Showalter, A.M., Keppler, B.D., Lichtenberg, J., Gu, D. and Welch, L.R. (2010) A bioinformatics approach to the identification, classification, and analysis of hydroxyprolinerich glycoproteins. Plant Physiology, 153, 485-513. doi:10.1104/pp.110.156554
- Spalding, J.D. and Hoyle, D.C. (2005) Accuracy of string kernels for protein sequence classification. Lecture Notes in Computer Science, 3686, 454-460.
- Zaki, N.M., Deris, S. and Illias, R. (2005) Application of string kernels in protein sequence classification. Applied Bioinformatics, 4, 45-52.
- Vries, J., Munshi, R., Tobi, D., Klein-Seetharaman, K., Benos, P.V. and Bahar, I. (2004) A sequence alignmentindependent method for protein classification. Applied Bioinformatics, 3, 137-148. doi:10.2165/00822942-200403020-00008
- Heinkoff, S. and Heinkoff, J. (1994) Protein family classification based on searching a database of blocks. Genomics, 19, 97-107. doi:10.1006/geno.1994.1018
- Heinkoff, S. and Heinkoff, J. (1994) A protein family classification method for analysis of large dna sequences. Proceedings of the 27th Annual Hawaii International Conference on Systems Sciences, New York, 265-274.
- Schultz, C.J., Rumsewicz, M.P., Johnson, K.L., Jones, B.J., Gaspar, Y.M. and Bacic, A. (2002) Using genomic resources to guide research directions. The arabinogalactan protein gene family as a test case. Plant Physiology, 129, 1448-1463. doi:10.1104/pp.003459
- Bendtsen, J.D., Nielsen, H., von Heijne, G. and Brunak, S. (2004) Improved prediction of signal peptides: SignalP 3.0. Journal of Molecular Biology, 340, 783-795. doi:10.1016/j.jmb.2004.05.028
- Nielsen, H., Engelbrecht, J., Brunak, S. and von Heijne, G. (1997) Identification of prokaryotic and eukaryotic signal peptides and prediction of their cleavage sites. Protein Engineering, 10, 1-6. doi:10.1093/protein/10.1.1
- Eisenhaber, B., Wildpaner, M., Schultz, C.J., Borner, G.H., Dupree, P. and Eisenhaber, F. (2003) Gylcosylphosphatidylinositol lipid anchoring of plant proteins. Sensitive prediction from sequenceand genome-wide studies for Arabidopsis and rice. Plant Physiology, 133, 1691-1701. doi:10.1104/pp.103.023580
- Johnson, K.L., Jones, B.J., Schultz, C.J. and Bacic, A. (2003) Non-enzymic cell wall (glyco) proteins. In: Rose, J.K.C., Ed., The Plant Cell Wall, Blackwell Publishers, Oxford, 111-154.