Health
Vol.4 No.11A(2012), Article ID:24983,14 pages DOI:10.4236/health.2012.431174
Pharmacological strategies for Parkinson’s disease
![]()
Department of Molecular Neuropathology, Institute of Cellular Physiology, (UNAM) National Autonomous University of Mexico, Mexico City, Mexico; *Corresponding Author: drucker@servidor.unam.mx
Received 5 October 2012; revised 8 November 2012; accepted 14 November 2012
Keywords: Parkinson’s Disease; Pharmacological Strategies; Preventive Agents; Dopaminergic Agents; L-DOPA
ABSTRACT
Parkinson’s disease (PD) or Paralysis Agitans was first formally described in “An essay on the shaking palsy”, published in 1817 by a British physician named James Parkinson. In the late 1950’s, dopamine was related with the function of the corpus striatum, thus with the control of motor function. But it was not until 1967, when the landmark study of George C. Cotzias, demonstrated that oral L-DOPA, the precursor of dopamine metabolism, was shown to induce remission of PD symptoms, that the definitive association between the two was firmly established. However, later on L-DOPA treatment began to show a loss of effectiveness and demonstrated to induce a variety of undesirable effects, the most prominent being diskinesia. As a result of this, a variety of alternative or complementary pharmacological strategies have been developed. In this chapter we review the wide variety of strategies that have been used through time, which are geared toward reducing the most disabling symptoms of PD. We additionally make some suggestions as to which are the most promising ones.
1. HISTORICAL ASPECTS OF PARKINSON’S DISEASE
Parkinson’s disease (PD) or Paralysis Agitans was first formally described in “An essay on the shaking palsy”, published in 1817 by a British physician named James Parkinson. However, there are earlier descriptions of PD dating back to 425 BC of ancient Chinese sources [1] and it has also been described in traditional Indian texts which named PD as Kampavata in Ayurvedic literature, the ancient Indian medical system [2].
PD is the second most common neurodegenerative disease, it affects approximately 1% - 3% of the population worldwide and it has been estimated that in the year 2030 PD prevalence will be twofold [3]. PD patients have severe motor alterations including resting tremor, muscle stiffness, paucity of voluntary movements and postural instability, which makes it particularly difficult for them to perform their daily activities and self-care tasks. The motor alterations have been related to the loss of dopaminergic neurons in the substantia nigra pars compacta (SNc) that leads to a reduction of dopamine release in the striatum (caudate and putamen nuclei).
Historically, the first well-established treatment for Parkinsonian tremor was with anticholinergic agents, used by Jean-Martin Charcot in the nineteenth century. For the next 60 years, dopamine loss in the striatum was not considered to be related to pathogenesis of PD, and the treatment was psychic rehabilitation with antispasmodic drugs such as atropine, scopolamine and stramonium [4], in addition with antihistaminic drugs as the most useful therapeutic agents [5].
It was not until the early twentieth century, in 1910, that dopamine was first synthesized by George Barger and James Ewens. For the next 30 years little work was done concerning dopamine, until Peter Holtz discovered dopa decarboxylase, the enzyme that transforms levodopa (also called L-DOPA) to dopamine, which then provided a mechanism for understanding dopamine formation.
Dopamine was thought to be only an intermediate product for noradrenaline and adrenaline formation, without a function for signal transduction. This perspective was modified with the discoveries in the late 50 s of Arvid Carlsson and coworkers (1957) who showed that L-DOPA administration was accompanied by motor hyperactivity. In 1958, this research group showed that dopamine was present in the brain and it was depleted by reserpine injection and restored by L-DOPA administration. In 1959, Bertler and Rosengred mapped dopamine distribution in the dog brain. They observed that the sites with the highest dopamine content contained little noradrenaline and that dopamine expression was prominent in the corpus striatum. This result was replicated in humans by Sano and coworkers [6]. Together, these findings led Bertler and Rosengred [7] to conclude that “dopamine is concerned with the function of the corpus striatum and thus with the control of motor function” suggesting that PD could be related to dopamine.
In 1960, Ehringer and Hornykiewicz [8] observed that striatal dopamine was depleted in the brain of PD patients but not in patients with Huntington’s chorea or extrapyramidal disorders. This discovery led Birkmayer [9] to perform the first pharmacological trial with LDOPA when they showed that intravenous injection of L-DOPA produced a transient antikinetic effect in these patients, leading the beginning of the era of L-DOPA therapy. However, it was not until the landmark study of George C. Cotzias in 1967 [10] who showed that low doses of oral L-DOPA, which was progressively increased over time, induced remission of PD symptoms. Cotzias not only reported that L-DOPA was effective in treatment of PD, but that dextro-DOPA provided no therapeutic result, but contributed to DOPA’s toxicity [10].
In the past two decades, the approach to PD has been modified due to the observations that other types of symptoms are associated to the disease. Consequently, PD is no longer considered merely as a motor disorder, but is rather a systemic disease which includes non-motor symptoms such as olfactory disturbances, sleep dysfunction, gastrointestinal abnormalities, and mood disorders [11-16]. Some of these symptoms appear before to motor deficits, and is referred as the premotor phase of PD and as such has become a new target for treatment guidelines in order to help delay or prevent the neurodegenerative process through an early diagnosis [17].
Nowadays, two centuries after the first formal description of the disease, there is still no cure. The goal of the current available pharmacological and non-pharmacological treatments, are to control mainly the motor symptoms since they are the most prominent and disabling ones. Unfortunately, they do not prevent the neurodegenerative process. L-DOPA, the gold-standard treatment, is limited given that over time and as progression of the disease ensues, the drug becomes less effective and patients show severe side effects such as freezing or akinesias. Therefore, the need for newer and more effective treatments is subject today of extensive research.
2. PATHOLOGY OF PARKINSON’S DISEASE
Several studies have suggested that the classical motor features of PD are preceded by non-motor symptoms. The period when these signs arise have been referred to as the premotor phase of PD. Among the best known premotor symptoms are olfactory disturbances, sleep alterations such as rapid eye movements (REM) sleep behavior disorder, constipation and mood alterations [11- 14,16,18].
PD symptoms gradually progress over years and have been divided in six neuropathological stages according to Braak’s staging system (2004). Each stage is marked by the continual increase of neuronal cytosolic filamentous inclusions from protein aggregates called Lewy bodies (LBs) and enlarged aberrant neurites (Lewy neurites, LNs) in specific neuronal populations of particular regions of the nervous system [19-25].
LBs and LNs are composed of aggregates of normal, misfolded and truncated proteins, and ubiquitin enzymes [26]. Their main component is α-synuclein [27,28], a presinaptic nerve terminal protein [29]. This protein is present in many of the nerve cells, but under certain conditions in some neuronal types, the secondary structure of α-synuclein changes to β-sheet structure which tends to form aggregates between other misfolded α-synuclein and other molecules [27,28,30-32]. The causes for the vulnerability of certain neuronal types to develop the abnormal protein aggregations are still unclear, but there are some hypotheses related to dopamine oxidation.
3. OXIDATIVE STRESS IN PD
PD is a multifactorial disorder. A large body of evidence suggests that loss of redox regulation contributes to the development of PD as reactive oxygen species could be responsible for neuronal death since it promotes α-synuclein aggregation (Figure 1). However, the etiology of PD is still poorly understood.
In a normal physiology there is an oxidation-reduction balance. Antioxidant molecules are in equilibrium given by antioxidant systems developed to prevent oxidative damage. When this oxidation-reduction equilibrium is lost due to an excess of oxidants or a deficit in the antioxidant systems, the oxidative-stress state is reached and plays an important role in the development of many degenerative diseases like PD [33]. Oxidative-stress state is characterized by high levels of reactive species and free radicals which are unstable molecules due to an unpaired electron. These molecules are extremely reactive and promote oxidative reactions with other molecules such as lipids, proteins and DNA in order to become stabilized.
Free radicals and reactive species are essential for normal physiology having regulatory process in cells (e.g. activation of transcription factors involved in cellular differentiation and proliferation), however, they have to be maintained in equilibrium in order to prevent cell damage. For this reason the organisms possess an elaborate network of antioxidant systems which neutralize free radicals and reactive species maintaining an oxidationreduction balance. Antioxidant systems are divided in enzymatic and non-enzymatic [34] including superoxide dismutase, glutathione peroxidase, catalase and thioredoxin as enzymatic systems and vitamins, proteins and amino-acids as nonenzimatic systems. These systems can be endogenous and exogenous. Exogenous antioxidant agents have been used in order to prevent oxidative damage induced by an excess of oxidants and a deficit in the antioxidant systems.
Dopamine can induce oxidative stress as it posseses a catechol group which oxidizes easily by transferring an electron to oxygen, forming different reactive species like hydrogen peroxide, superoxide anion and hydroxyl radicals. These reactive species are able to generate an oxidative-stress state which when becoming chronic, inhibits the activation of endogenous antioxidant systems, making the organism unable to deal with the production of free radicals and reactive species.
Dopamine oxidizes to form dopamine o-quinone, a non-stable molecule in cytosol at physiological pH. The instability induces the amino chains to cyclize and to generate aminochrome [35]. Aminochrome rearrangement produces 5,6-dihydroxindole which is oxidized and then polymerizes to form neuromelanin [36] which can act as chelator for metals, playing a neuroprotective role [37,38]. On the other hand, aminochrome is able to participate in toxic reactions by interacting with complex I and III of mitochondria inducing an energy dysfunction and also interacting with α-synuclein to form protofibrils. Both have been implicated in cell death (Figure 1).
Neuromelanin forms a stable complex with ferric iron, the most abundant metal in human body including the brain. In PD patients a high accumulation of this metal has been observed in the SNc [39,40] suggesting that it could be causing the dopaminergic neuronal death as it induces oxidative stress by generating hydroxyl radicals from free irons (through Fenton reaction). In addition, iron can induce α-synuclein aggregation [41] as a possible consequence of the oxidative stress [42]. It has been suggested that iron released from neuromelanin increases oxidative stress in mitochondria causing an energy dysfunction in the cell, and also reduces proteosome function which is a hallmark of PD [43] (Figure 1).
4. PHASES OF PARKINSON’S DISEASE
4.1. Premotor Stage
Braak and coworkers [44] proposed that symptoms of PD evolve sequentially and advance in a predictable manner. According to Braak, the disease begins with neurophatological changes (LBs and LNs) observed in the olfactory bulb and the medulla oblongata. The former has been related to the olfactory disturbance present at early stages of PD. Olfactory deficit is one of the most well known premotor symptoms. It has been reported that hyposmia, which is the loss of smell capacity, is present in over 80% of PD patients [13]. Constipation, another well known premotor symptom, can be associated with the degeneration of the dorsal nucleus of the vagus nerve in stage 1 [14]. Then, at stage 2, degeneration of locus coeruleus and pedunculo pontine nucleus begins to appear. These structures have been related with the neuropathological substrates of REM sleep disorder, however it is not definitively established [45-47].
4.2. Motor Stage
The motor symptoms of PD are associated to the degeneration of dopaminergic neurons in the SNc, with the subsequent loss of dopamine neurotransmission in the striatum (stage 3 in Braak staging system). The striatum is the major input nucleus of the basal ganglia (BG) which consists of a complex network that integrates cortical areas, basal ganglia nuclei and thalamus [48].
In the late 1980s, Albin [49] and DeLong [50] proposed a model to explain the organization of the BG and how dopamine loss leads to motor disturbances in PD. Anatomically, the functional model of the BG, suggests that there are two main pathways originating in the striatum that control motor behavior: the direct pathway and the indirect pathway. In both pathways, GABAergic medium spiny neurons (MSNs, or projection neurons, the main neuronal type within the striatum) receive glutamatergic inputs from the cerebral cortex and thalamus, and dopaminergic inputs from SNc. MSNs from the direct pathway project directly to substantia nigra pars reticulata (SNr, GABAergic neurons) and globus pallidus interna (GPi, GABAergic neurons), which are the output nuclei of the BG. MSNs from the indirect pathway project to external globus pallidus (GABAergic neurons) that projects to subthalamic nucleus (glutamatergic neurons) and finally to SNr/GPi. GABAergic neurons from SNr/GPi project directly to the thalamus, which contains glutamatergic neurons, that project to the cerebral cortex.
Dopamine has been known to play a critical role in modulating excitation of MSNs through D1 and D2 dopamine receptors (D1R and D2R respectively). Dopamine receptors are classified in two classes: D1-class and D2-class, which are G-protein-coupled receptors. D1R belongs to the D1-class dopamine receptors that activate Gαs/olf family and stimulated cAMP production by activation of adenylyl cyclase. D2R belongs to the D2-class dopamine receptors that are coupled to the Gαi/o family and induce inhibition of adenylate cyclase. Therefore, activation of D1R increases striatal output, whereas activation of D2R reduces striatal output [49-51].
In addition to the opposing responses of DR, it has been proposed that their expression is segregated within the two pathways [52,53]. Recently, this hypothesis has been confirmed by Kravitz and coworkers [53] showing that D1R is preferentially expressed in neurons of the direct pathway, whereas D2R is preferentially expressed
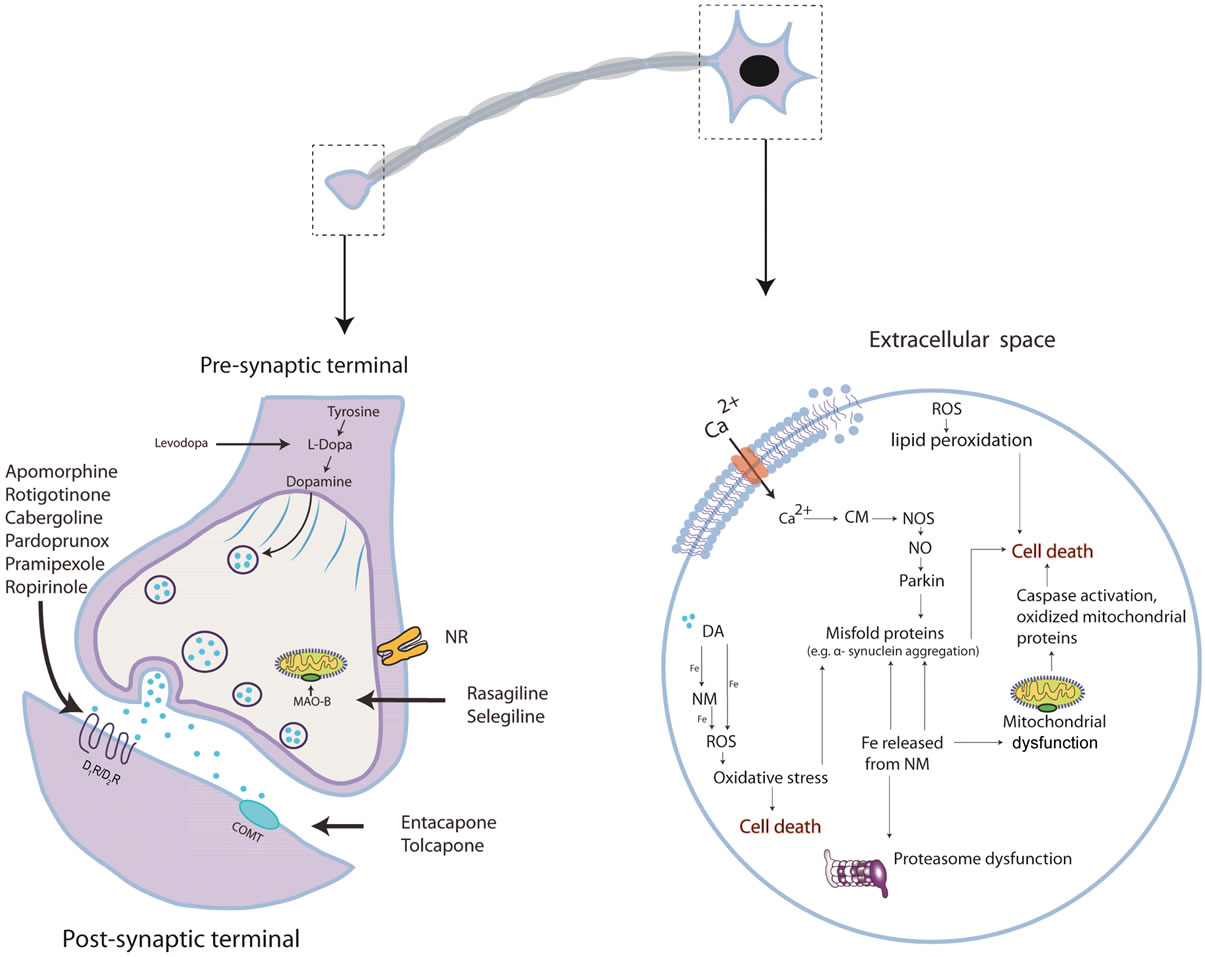 (a) (b)
(a) (b)
Figure 1. (a) Representative scheme of the current drugs used to treat PD; Pardoprunox, Pramipexole, Rotigotine, Ropirinole and Carbergilone D1/D2 agonists used to avoid dyskinesias; COMT inhibitors to avoid the degeneration of dopamine; MAO-B inhibititors to avoid the presinaptic dopamine degradation; the pre-synaptic nicotinic receptor to release dopamine and the post-synaptic nicotinic receptor which sensitizes dopamine recepror; (b) Dopaminergic neuronal death in PD is not well understood. It involves at least protein misfolding and α-synuclein aggregation. The ubuquitin-proteasome system and Parkin (related with genetic PD) degrades these damaged proteins. In PD, oxidative stress may play a crucial role in cell death as α-synuclein aggregation can be toxic by forming reactive oxygen spices (ROS) that can induce different types of damage: impaired ubiquitin-proteasome system and oxidizing Parkin leading to accumulation of protein aggregates. Mitochondrial dysfunction is another suggested pathway which induces cell death. Impaired oxidative phosphorilation in mitochondrial membrane and decreased complex I activity which produces the formation of ROS has been observed. Mitochondrial membrane alteration can result in the release of cytochrome C to the cytosol activating pro-apoptotic pathways as caspase. Ca2+ influx to the cell can results in nitric oxid (NO) production; Ca2+ binds to calmodulin (CM) activating nitric oxide synthase (NOS) which produces NO. NO can generate the free radical peroxynitrite (OONO) by interacting with superoxide anion radical (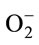 ). NO can also alter protein folding and as in PD there is a proteasome dysfunction and Parkin impairment due probably to the oxidative stress state and thus cannot be degraded. It has been suggested that dopamine can also induce the formation of ROS; dopamine released from the vesicles to the cytosol is able to coordinate Fe to form neuromelanin (NM) and ROS. NM also can coordinate Fe and produces ROS. Membrane lipid peroxidation is another consequence of oxidative stress which produces membrane instability and finally cell death.
). NO can also alter protein folding and as in PD there is a proteasome dysfunction and Parkin impairment due probably to the oxidative stress state and thus cannot be degraded. It has been suggested that dopamine can also induce the formation of ROS; dopamine released from the vesicles to the cytosol is able to coordinate Fe to form neuromelanin (NM) and ROS. NM also can coordinate Fe and produces ROS. Membrane lipid peroxidation is another consequence of oxidative stress which produces membrane instability and finally cell death.
in neurons of the indirect pathway.
Functionally, activation of direct or indirect pathway has different outcomes in motor behavior. It has been proposed that stimulation of the direct pathway facilitates movement, whereas stimulation of the indirect pathway suppresses motor behavior [49,50]. This hypothesis has been demonstrated by in vivo activation of one of the two pathways, with an optogenetic approach utilizing Credependent viral expression of chanelrhodopsin-2 in BAC transgenic D1- or D2-driven Cre lines [53].
Motor control has long been thought to be achieved through the balanced activity of the two pathways. In agreement with this hypothesis, in PD there is an imbalance of these pathways as dopamine modulates directand indirect-pathway activation. This imbalance results in motor alterations (deficits in movement initiation and execution) owing to dopamine depletion into the striatum.
4.3. Late Stage
Motor disturbances appear when the pathological process infiltrates the midbrain and forebrain at stage 4. Once this happens, the progression rate of the disease differs depending on the age of symptom onset [54] with further infiltrations of LBs and LNs into more brain regions such as amygdala, medial temporal cortex, and higher order association cortices and finally into primary cortices.
According with Braak, stages 5 and 6 implicate that neurodegeneration proceeds to first order sensory association areas, premotor and high-order sensory association areas, prefrontal neocortex, and finally into primary sensory and motor areas. This degeneration induces several symptoms including hallucinatory episodes, dementia and states of confusion. Dementia in PD has been correlated with atrophy of nucleus basalis Meynert which results in a cholinergic deficit.
5. PHARMACOLOGICAL STRATEGIES
Even though motor alterations in PD appear long after the disease begins, these symptoms are the main target of pharmacological treatment. L-DOPA, the gold standard treatment, loses its effectiveness after several years and involves severe undesirable effects like dyskinectic movements, thus it is important to delay L-DOPA use as long as possible in order to extend its positive effects and delay the adverse effects. Additionally, in order to modify this therapeutic approach, researchers have emphasized the importance of an early diagnosis through premotor signs of PD, in order to attempt to prevent or delay the neurodegenerative process or at least the appearance of motor signs.
In the next part of the review we will discuss different treatments currently used in accordance with the progression of the disease. Nowadays, the therapeutic approach of PD is poor due to the limited knowledge about the physiopathology of the disease.
5.1. Preventive Agents
The main target of preventive agents is to delay the neurodegenerative process. As we described previously, it has been suggested that neuronal death in PD is associated to a chronic loss of redox regulation that leads the activation of diverse cell damage processes.
5.1.1. Antioxidants
Exogenous antioxidants such as vitamins C, D and E are able to donate electrons to free radicals neutralizing them in order to prevent oxidation. Vitamins C and E are not preventive antioxidants, they act as switches stopping the formation of free radical in chain reactions [34]. Vitamin C also traps peroxyl radicals and inhibits lipid peroxidation which leads to cell membrane alterations [55]. Even though these vitamins do not cross the blood brain barrier they have been administrated in PD patients to prevent the progression of the disease, delaying the beginning of L-DOPA therapy for 2.5 to 3 years [56,57].
A growing body of evidence suggests that vitamin D therapy may be beneficial for PD patients. Vitamin D have been related to PD as it has an important role in different processes including cellular metabolism, oxidative stress, inflammation, gene expression among others altered in PD (Figure 1). Furthermore, vitamin Dknockout mice showed motor deficits, making Vitamin D receptor a neuronal biomarker candidate for PD [58].
Other exogenous antioxidants are isoflavonoids, like those contained in soja, specifically genistein and dadzein. Isoflavonoids are plant secondary metabolites which can exert hormonal and non-hormonal properties, with antioxidant effects given by their reductive capacity, which can be a possible mechanism whereby they protect against chronic diseases. Another possible protective effect is that isoflavonoids reduces the massive calcium entry to the cell and inhibits pro-apoptotic pathway activation [59]. These antioxidant agents diminish the inflammatory pathway activation, stabilizing mitochondrial integrity after harmful stimuli.
Epidemiologic studies have shown that the rate incidence is higher in people who did not incorporate the fatty acid to the daily diet [60]. Decosahexaenoic acid (DHA) is the most abundant Omega-3 fatty acid in the brain. This molecule has different effects in cell signaling pathways. It has been shown that this compound increases the anti-apoptotic signaling cascade by the apoptotic cell agonist BCL-2 [61,62].
Green tea which is widely used in Asia, is another antioxidant agent. This bevarage has been related to a low incidence of cardiovascular alterations, cancer and PD in that population. Epidemiological studies from China have shown a minor incidence of PD when compared to Caucasians [63,64]. The underlying protective mechanisms can be explained by the polyphenols contained in the tea, as they are a 100 fold more powerful as antioxidant than vitamin C and 25 fold more than vitamin D [65]. In addition, green tea as well as coffee contains caffeine, an adenosine A2A blocker which facilitates dopaminergic action in the striatum [65].
5.1.2. Iron Chelators
Considering the evidence of iron role in oxidative stress and cellular damage, a therapeutic approach is the administration of iron chelators. These molecules have been shown to have neuroprotective effects [66] by sequestering the free iron preventing the formation of hydroxyl radicals [67].
Several iron chelators like VK-28 [5-(4-(2-hydroxyethyl) piperazin-1-yl (methyl)-8-hydroxyquinoline] and M30 [5-(N-methyl-N-propargyaminomethyl)-8-hydroxyquinoline], designed to cross the brain-blood-barrier [68], have been shown to reduce the neuronal death caused by oxidative stress in animal models of PD. Thus, iron chelators could be a potential target for PD preventive therapy.
5.1.3. Caffeine
Epidemiological studies show a minor incidence rate of PD in regular coffee drinkers suggesting a neuroprotective effect [69,70]. Coffee is one of the beverages used worldwide that contains caffeine, which is an antagonist of adenosine receptors that has been widely studied as a neuroprotective agent. However, it has been observed that decaffeinated coffee may also protect against damage, making controversial the interpretation of the epidemiological studies [71]. In any case the mechanism by which coffee offers protection to the central nervous system (CNS) is poorly understood. Some studies have conferred the neuroprotective effect to antagonism toward the A2A adenosine receptor. This may reduce the negative effects of interleukine 1b, diminishing the proapoptotic cascade [72]. On the other hand it has been reported that antagonists of A2A receptor can inhibit the glutamate release in the subtantia nigra pars compacta, diminishing the neurotoxicity through calcium entry to the dopaminergic neurons [73]. In contrast, the beneficial effect of A2A antagonists could depend on their ability to tune the indirect pathway. Future studies could focus on evaluating the role of A2A receptors on the striatal circuitry as a potentially relevant therapeutic candidate (Figure 2).
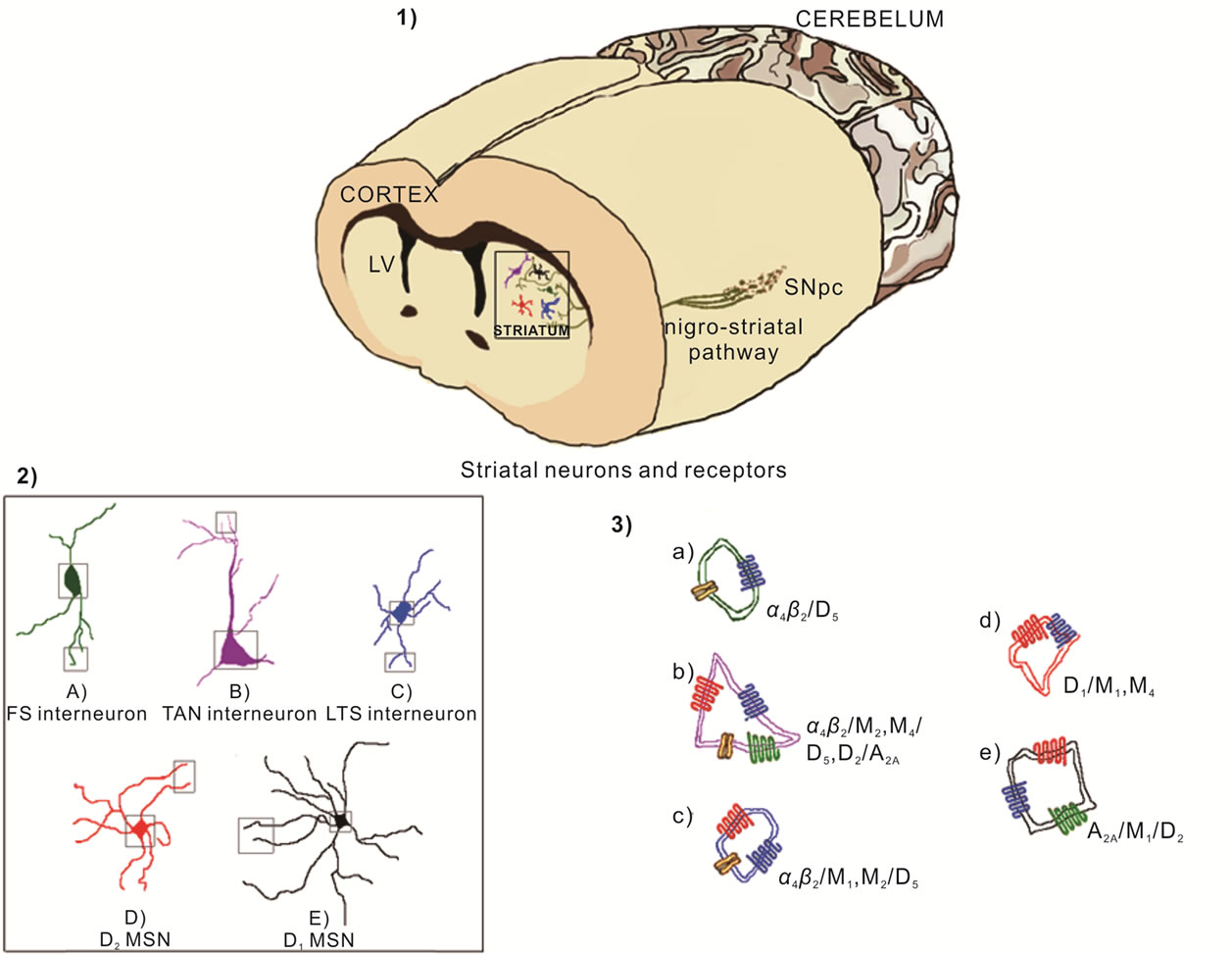
Figure 2. Nigrostriatal pathway transmits dopamine from substance nigra pars compacta (SNpc) to the striatum (1) The striatum has different neuronal types including the projection neurons named medium spiny neurons (MSN) and fast spiking (FS) interneurons, low threshold spiking (LTS) interneurons and tonically active cholinergic neurons (TAN); MSN can be classified by the expression of dopamine receptors as D1-MSN and D2-MSN (2); These neurons express both types of cholinergic receptors as nicotinic receptor like α4β2 and muscarinic receptor like M1, M2 and M4, as well as adenosine receptors (A2A) and dopaminergic receptors (D1, D2 and D5) (3).
5.2. Treatment Agents
5.2.1. L-DOPA the “Gold Standard” Treatment
L-DOPA (L-3,4 dihydroxiphenilalanine) is the gold standard treatment of PD, used in stages when motor symptoms begin to be present. All current L-DOPA products are formulated with aromatic amino acid decarboxylase inhibitors to prevent the metabolism of levodopa in the gastrointestinal tract and systemic circulation. The absortion of L-DOPA in the digestive tract is via the saturable L-neutral amino acid transport system for large amino acids. Levodopa crosses the blood-brain barrier by stereospecific, saturable, facilitated process via the large neutral aminoacid (LNAA) transport carrier system. As it has been documented, the primary complication of L-DOPA therapy is the development of dyskinesias and motor fluctuations. Dyskinesias have a variety of clinical forms, including dystonic and choreic movements that occur in different points of L-DOPA cycle. The most common forms of dyskinesias are the “OFFperiod dystonia” the “diphasic dyskinesias” (onset and end of dose dyskinesias) and the “peak dose dyskinesias”. Motor fluctuations are divided in two periods with opposite effects. “On” period when patients are without motor symptoms, and the “Off” period when L-DOPA lose its effectiveness and motor symptoms return. L-DOPA complications have been related with the diminished number of dopaminergic neurons, since the ability to transform L-DOPA to dopamine is altered with the progression of dopamine degeneration. Although, it has been reported that serotonergic terminals metabolize L-DOPA to dopamine by L-Aromatic aminoacid descarboxilase (LAAAD) enzyme, but this metabolism is insufficient in late stages of PD. Dyskinesias have been conferred to an altered dopamine receptor. Many pharmacological strategies have been employed to avoid the disabling symptoms of dyskinesias. In order to improve the quality of life, specific D2-D3 dopamine receptor agonists have been used. Considering the side effects of L-DOPA therapy, the dose in combination with carbidopa, has to be adjusted in accordance with patient’s response, ranging from 125/12.5 mg up to 800/200 mg. Carbidopa is a peripheral dopamine decarboxilase inhibitor that does not cross the brain barrier. The maximum dosage of L-DOPA/Carbidopa is 1500/200 mg divided in three doses per day. L-DOPA can also be mixed with benzerazide, starting with 100/25 mg per day.
5.2.2. IMAO B Inhibitors
There are two types of mono-amino-oxidase (MAO) enzymes in humans; A and B. MAO-A is mainly located in the intestine whereas MAO-B is preferentially expressed in the CNS. As MAO regulates the releasable stores and free cellular levels of dopamine, their inhibition is therapeutic in order to increase dopamine levels in the brain.
Selegiline is an irreversible inhibitor of MAO-B. Selegiline has proved to increase the levels of amines in the striatum and was the first selective inhibitor used to treat PD avoiding the side effects related to cheese intake when the patients are on IMAO-A therapy (increased blood pressure by high cheese intake) [74]. The treatment with selegiline is used at doses that range from 4 - 8 mg/day. It has been reported that selegiline also increase the levels of growth factors like BDNF and GDN, which opens the possibility to stop the neuronal loss in the SNc [75].
Rasagiline, a selective inhibitor of MAO-B, is 5-fold more potent than selegiline following repeated administrations. In contrast to selegiline, rasagiline is not metabolized to amphetamine, event which increases the striatal extracellular dopamine levels. Rasagiline also has demonstrated to have neuroprotective effects in animal models of PD by activating anti-apoptotic genes [76]. The recommended dosage based in the ADAGIO (Attenuation of disease progression with “azilect” rasegiline given once daily) study is around 0.5 and 1 mg/day.
5.2.3. COMT Inhibitors
One of the most important targets of L-DOPA therapy for PD is to prolong the duration of antiparkinsonian effects without an increase peak on plasma concentration in order to provide a longer benefit without disabling diskinesias. Cathecol 0-Methyl Transferase(COMT) is an enzyme that degrades L-DOPA. Thus blocking COMT activity with inhibitors like entacapone to avoid their degradation enhances the L-DOPA re-uptake and dopamine transformation.
Entacapone and tolcapone are the most used COMT inhibitors. They have shown to reduce “Off” periods for one hour, and are recommend in advanced PD. Nowadays, tolcapone is no longer used due to the hepatotoxicity that it produces. Entacapone dosage range used to treat PD is 200-2000 mg in order to reduce by 10% - 30% L-DOPA dosage [77].
5.2.4. Dopaminergic Agonists
Dopaminergic agonists could be divided in two groups: ergolines which are derivates from ergot alkaloids and includes bromocriptine, pergolide, lisuride, alphadihydroergocryptine and cabergoline; and non-ergolines like apomorphine, pramipexole, popirinole, potigotine and pardoprunox, among others. Most of the non-ergolines have been discovered recently, with the exception of apomorphine.
Pramipexole is a -D2, D3, D4-receptor agonist. The effective dosage range of pramipexole in early stages of PD is 0.375 to 4.5 mg/day. It has two pharmacological presentations; the immediate release type and the extended release type [78]. Although pramipexole has shown to induce diminished motor symptoms associated with L-DOPA, it has several side effects including hallucinations, edema, excessive diurnal somnolence, and impulse control disorders like pathologic gambling and hypersexuality [56].
Ropinirole is a -D2, D3, D4-receptor agonist ameliorates PD symptoms. Effective doses in early PD range between 8 to 12 mg/day with a half life of 6 to 8 hours. It has been reported that ropinirole has the same side effects as pramipexole and ropinirole but is less effective than L-DOPA for motor symptom improvement. Nevertheless it reduces dyskinesias [79].
Rotigotine is a -D1, D2, D3-receptor agonist which is a lipid-soluble derivate from aminotetraline, nowadays available as transdermal patch with a lower risk of accumulation due to its 5 - 7 hour half life. Rotigotine has been tested in early motor-stages of PD, showing no differences between the pramipexole and ropinirole at dosages that ranges from 9 to 13.5 mg per day. The transdermal application of rotigotine is appropriate in patients who are not able to swallow. Another positive feature is that the dosage of rotigotine by transdermal patch is maintained constant during 24 hours, providing a lower risk to develop motor fluctuations as has been proven in “The Parkinson study group” [80].
Pardoprunox (SLV308) is a partially dopamine D2- and D3-receptor agonist and a full 5HT(1A) serotonin agonist that has been recently approved by FDA for PD treatment. Experiments developed in animal models of PD showed that pardoprunox has beneficial motor effects [81]. Pardoprunox also showed to be a good agonist of D2-dopamine receptor in the VTA reducing the burst activity linked to the phasic activity of dopaminergic neurons. Studies in PD patients showed that pardoprounox mixed with L-DOPA can decrease the motor fluctuations and has anti-depressive properties [82].
Apomorphine is a non selective dopamine agonist injected via subcutaneous to avoid the first-pass metabolism by the liver. This drug is also called the rescue therapy as it is effective during the next hour of injection of 30 mg. It is considered an ideal therapy for patients with advanced stages of PD and painful off-periods and also recommended for special situations in which it is inconvenient to have an off-period when being away from home [83].
Cabergoline is a synthetic ergoline derivate with a high and prolonged affinity for D2- and D3- dopamine receptors, and lower affinity for D1 receptors. Cabergoline also has been demonstrated to have a little affinity to serotonergic and adrenergic receptors. It has shown to have neuroprotective effects in animal models of PD by reducing lipid peroxidation and the ability to scavenge free radicals [84]. The half life of cabergoline is 24 hours and is given to PD patients at range of 2 to 6 mg/day to achieve motor improvement [85]. Studies in patients with early idiopathic PD in a single 5 year trial have suggested that cabergoline could also be given as monotherapy at early stages of PD [86].
5.2.5. Anticholinergic Therapy
Anticholinergic agents were the first therapies for PD. It is well established that the decrease of striatal dopaminergic innervation increases cholinergic activity in the striatum. The stability between DA and Ach in PD patients is disrupted increasing the cholinergic activity, suggesting a dopamine-ACh imbalance may underlie motor symptoms of PD [87]. It should be noted that older PD patients do not tolerate anticholinergic drugs, thus they are recommended only in younger patients.
Amantadine remains the only drug to treat PD diskynesias induced by L-DOPA [88]. Amantadine is a 5HT antagonist and a dopamine reuptake blocker. The dosage range is 300 - 400 mg/day. However, amantadine has several side effects including confusion, dry mouth, constipation, blurred vision, urinary retention and cognitive impairment, although it is substantially less potent than dopaminergic agonists [89].
5.2.6. Nicotine
Several epidemiologic studies have shown an inverse correlation between smoking and PD in case-control, open-population and cohort studies. This suggests that nicotine may have neuroprotective effects and therefore could be preventing PD [90-94].
Nicotine neuroprotection has been demonstrated in cell culture experiments and animal models of neurodegenerative diseases. In vitro, nicotine protects against toxicity mediated by glutamate, arachidonic acid and others toxic compounds [95-97] including the neurotoxin MPTP [96]. In the rat model of PD, nicotine protects against 6-OHDA [97-99] and also protects against MPTP in mice and non-human primates models [100,101]. Although nicotine administration has been reported to protect against cell damage [97,102], there are, however, some contrary results showing no protection even under chronic nicotine administration [71,103]. These conflicting results could be resolved by attempting to understand nicotine action in subjects with nigral degeneration. As early as 1926 Moll reported that in patients with postencephalitic Parkinsonism, nicotine reduced motor symptoms. More recently, nicotine administered in different forms (gum and patch; low and high doses) has resulted in a reduction of PD symptoms [104-106]. The symptoms relieved by nicotine have also been studied in animal models of PD, where it has been reported that it prevents the akinesia and rigidity developed by L-DOPA therapy [107]. This indicates that nicotine has significant effects, in striatal circuitry, where nicotine acetylcholine receptors (NR) are ubiquitously present in striatal neurons [108] (Figure 2). Therefore it is conceivable that nicotine action rather than being protective, may instead be involved in modifying basal ganglia activity in such a way that it produces some significant and beneficial effects on motor disruptions produced by PD. Therefore the protective effect may be physiological rather than anatomical.
The following processes may therefore be involved. Striatum is part of BG and has been strongly implicated in motor behavior. Striatal circuitry is a complex system that is 95% composed of MSNs [109]. These neurons are classically classified in relation to their postsynaptic targets in the direct pathway, which expresses D1 receptors, which facilitates movement and in the indirect pathway, which expresses D2 receptors, and depresses movement (Figure 2). The MSNs activity (direct/indirect) is modulated by interneurons, which represent the other 5% of striatal neuronal population. Inter-neurons are classified by their spike pattern, which are fast (FS) and low threshold (LTS) both of them are GABAergic (Learn and Grafstrom, 1989). In addition the striatum has tonically active cholinergic neurons (TAN) [110]. Interneurons respond to nicotine since they have NR. Fast spiking interneurons has 100% of nicotinic current, tonically active neurons has 44% and low threshold 33% [111] (Figure 2). According to this approach, it has been observed that blocking FS activity induces diskinesias [112] on the other hand NR activates these neurons, highlighting the possible pharmacological use of nicotine agonists to treat complications of PD. Taking into consideration that PD is progressively injuring dopaminergic neurons, chronic stimulation of NR may chronically stimulate dopamine release during this damage. Therefore nicotine could progressively modify dopaminergic dynamics by increasing the sensitivity or abundance of DA receptors [113,114]. Other neurotransmitters could importantly be afected by nicotine, like the gammaaminobutyric acid (GABA) [115], as well as glutamate [116], serotonin [117] and opioid peptide [118,119]. In example of regulation, the hypercholinergic state observed in the PD model could be decreased by nicotine [114,120]. Another example could be the cortico-striatal glutamatergic presynapses that have the α7 NR and also promotes the glutamate release, the main neurotransmitter involved in long term potentiation and long term depression phenomena.
Altogether these findings open the possibility to use specific nicotine agonists, in attempting to modulate the output signal of striatum, by modifying the firing pattern of GABAergic striatal interneurons [121]. The classic scheme of the basal ganglia function suggests that imbalance between direct and indirect pathway promotes the movement symptoms of Parkinson’s disease. Nicotine has an important effect on the direct pathway, in contrast with other drugs which only have effects on the indirect pathway. This makes nicotine an interesting therapeutic strategy in the treatment of PD [122].
6. CONCLUSIONS
Great pharmacological efforts have been done to treat PD, nevertheless it has been insufficient due to motor complications which tend to arise as the disease progresses.
Current strategies to treat PD are focused on early stages in order to prevent neuronal degeneration and motor disturbances, thus the use of bio-markers to identify the onset of the pathology has shown promissory applications.
As motor symptoms become clinically detectable, PD has to be treated with circuitry modifiers in order to avoid dyskinesias and motor fluctuations. Current drugs, like the non selective DA agonist with serotoninergic action, have induced a better response than DA agonists only.
Striatal adenosine and nicotine receptors (A2A and a4b2, respectively) are promissory targets to treat PD, therefore, these drugs may currently represent good and important strategies to help diminish the undesirable symptoms which frequently appear after prolonged drug treatment.
7. ACKNOWLEDGEMENTS
We would like to thank Marcela Palomero-Rivero, Diana MillánAldaco, Francisco Pérez-Eugenio for their excellent technical assistance. This work was supported by the Project Program Grant IMPULSA-UNAM 02 and UNAM-DGAPA-PAPIIT grants IN204612 to R. Drucker-Colín. JRGM and ABG received a Ph.D. fellowship from CONACYT-México.
![]()
![]()
REFERENCES
- Zhang, Z.X., Dong, Z.H. and Roman, G.C. (2006) Early descriptions of Parkinson disease in ancient China. Archives of Neurology, 5, 782-784. doi:10.1001/archneur.63.5.782
- Manyam, B.V. (1990) Paralysis agitans and levodopa in “Ayurveda”: Ancient Indian medical treatise. Movement Disorders, 1, 47-48. doi:10.1002/mds.870050112
- Dorsey, E.R., Constantinescu, R., Thompson, J.P., Biglan, K.M., Holloway, R.G., Kieburtz, K., Marshall, F.J., Ravina, B.M., Schifitto, G., Siderowf, A. and Tanner, C.M. (2007) Projected number of people with Parkinson disease in the most populous nations, 2005 through 2030. Neurology, 5, 384-386. doi:10.1212/01.wnl.0000247740.47667.03
- Vollmer, H. (1942) Comparative value of solanaceus alkaloids in treatment of Parkinson’s syndrome. Archives of Neurology and Psychiatry, 48, 72-84.
- Meter, J.R.V. (1950) Therapy of Parkinson’s disease. California Medicine, 4, 322-324.
- Sano, I., Gamo, T. and Kakimoto, Y. (1959) Distribution of cathecol compounds in human brain. Biochemica et Biophysica Acta, 32, 586-587. doi:10.1016/0006-3002(59)90652-3
- Bertler, A. (1959) Ocurrence and distribution of dopamine in brain and other issues. Experientia, 15, 10-11. doi:10.1007/BF02157069
- Hornykiewicz, O. and Ehringer, J. (1960) Verteilung von noradrenalin and dopamin im gehirn des menschen und ihr verhalten bei erkrankungen des extrapyramidalen systems. Klinische Wochenschrift, 38, 1126-1239.
- Birkmayer, W. and Hornykiewicz, O. (1961) The L-3,4- dioxyphenylalanine (DOPA)-effect in Parkinson-akinesia. Wiener Klinische Wochenschrift, 73, 787-788.
- Cotzias, G.C., Van Woert, M.H. and Schiffer, L.M. (1967) Aromatic amino acids and modification of parkinsonism. The New England Journal of Medicine, 7, 374-379. doi:10.1056/NEJM196702162760703
- Ansari, K.A. and Johnson, A. (1975) Olfactory function in patients with Parkinson's disease. Journal of Chronic Diseases, 9, 493-497. doi:10.1016/0021-9681(75)90058-2
- Doty, R.L., Golbe, L.I., McKeown, D.A., Stern, M.B., Lehrach, C.M. and Crawford, D. (1993) Olfactory testing differentiates between progressive supranuclear palsy and idiopathic Parkinson’s disease. Neurology, 5, 962-965. doi:10.1212/WNL.43.5.962
- Doty, R.L. (2012) Olfactory dysfunction in Parkinson disease. Nature Reviews Neurology, 6, 329-339.
- Del Tredici, K., Rub, U., De Vos, R.A., Bohl, J.R. and Braak, H. (2002) Where does parkinson disease pathology begin in the brain? Journal of Neuropathology & Experimental Neurology, 5, 413-426.
- Postuma, R.B. and Montplaisir, J. (2009) Predicting Parkinson's disease—why, when, and how? Parkinsonism & Related Disorders, 15, S105-S109. doi:10.1016/S1353-8020(09)70793-X
- Ravina, B., Camicioli, R., Como, P.G., Marsh, L., Jankovic, J., Weintraub, D. and Elm, J. (2007) The impact of depressive symptoms in early Parkinson disease. Neurology, 4, 342-347. doi:10.1212/01.wnl.0000268695.63392.10
- Schapira, A.H. (2010) Future strategies for neuroprotection in Parkinson's disease. Neurodegenerative Disease, 1-3, 210-212. doi:10.1159/000295666
- Postuma, R.B., Gagnon, J.F., Vendette, M. and Montplaisir, J.Y. (2009) Markers of neurodegeneration in idiopathic rapid eye movement sleep behaviour disorder and Parkinson’s disease. Brain, Pt 12, 3298-3307. doi:10.1093/brain/awp244
- Forno, L.S. (1996) Neuropathology of Parkinson’s disease. Journal of Neuropathology & Experimental Neurology, 3, 259-272. doi:10.1097/00005072-199603000-00001
- Jellinger, K.A. (2003) Alpha-synuclein pathology in Parkinson’s and Alzheimer’s disease brain: Incidence and topographic distribution—A pilot study. Acta Neuropathologica, 3, 191-201. doi:10.1007/s00401-003-0725-y
- Ohama, E. and Ikuta, F. (1976) Parkinson’s disease: Distribution of Lewy bodies and monoamine neuron system. Acta Neuropathologica, 4, 311-319. doi:10.1007/BF00696560
- Braak, H., Del Tredici, K., Bratzke, H., Hamm-Clement, J., Sandmann-Keil, D. and Rub, U. (2002) Staging of the intracerebral inclusion body pathology associated with idiopathic Parkinson’s disease (preclinical and clinical stages). Journal of Neurology, 3, 1-5. doi:10.1007/s00415-002-1301-4
- Dickson, D.W., Braak, H., Duda, J.E., Duyckaerts, C., Gasser, T., Halliday, G.M., Hardy, J., Leverenz, J.B., Del Tredici, K., Wszolek, Z.K. and Litvan, I. (2009) Neuropathological assessment of Parkinson’s disease: Refining the diagnostic criteria. The Lancet Neurology, 12, 1150- 1157. doi:10.1016/S1474-4422(09)70238-8
- Lewy (1919) Paralysis agitans. I. pathologische anatomie. In: Lewandowsky, M., Ed., Hundbuch der Neurologie, 920-933.
- Tretiakoff (1919) Contribution a L’étude de L’anatomie Pathologique de Locus Niger de Soemmerling. Université de Paris, Paris.
- Xia, Q., Liao, L., Cheng, D., Duong, D.M., Gearing, M., Lah, J.J., Levey, A.I. and Peng, J. (2008) Proteomic identification of novel proteins associated with Lewy bodies. Frontiers in Bioscience, 13, 3850-3856. doi:10.2741/2973
- Wakabayashi, K., Takahashi, H., Obata, K. and Ikuta, F. (1992) Immunocytochemical localization of synaptic vesicle-specific protein in Lewy body-containing neurons in Parkinson's disease. Neuroscience Letters, 2, 237-240. doi:10.1016/0304-3940(92)90923-U
- Spillantini, M.G., Schmidt, M.L., Lee, V. M., Trojanowski, J.Q., Jakes, R. and Goedert, M. (1997) Alpha-synuclein in Lewy bodies. Nature, 6645, 839-840. doi:10.1038/42166
- Iwai, A., Masliah, E., Yoshimoto, M., Ge, N., Flanagan, L., de Silva, H.A., Kittel, A. and Saitoh, T. (1995) The precursor protein of non—A beta component of Alzheimer’s disease amyloid is a presynaptic protein of the central nervous system. Neuron, 2, 467-475. doi:10.1016/0896-6273(95)90302-X
- Baba, M., Nakajo, S., Tu, P.H., Tomita, T., Nakaya, K., Lee, V.M., Trojanowski, J.Q. and Iwatsubo, T. (1998) Aggregation of alpha-synuclein in Lewy bodies of sporadic Parkinson’s disease and dementia with Lewy bodies. American Journal of Pathology, 4, 879-884.
- Hashimoto, M., Hsu, L.J., Xia, Y., Takeda, A., Sisk, A., Sundsmo, M. and Masliah, E. (1999) Oxidative stress induces amyloid-like aggregate formation of NACP/alphasynuclein in vitro. Neuroreport, 4, 717-721. doi:10.1097/00001756-199903170-00011
- Fujiwara, H., Hasegawa, M., Dohmae, N., Kawashima, A., Masliah, E., Goldberg, M.S., Shen, J., Takio, K. and Iwatsubo, T. (2002) alpha-Synuclein is phosphorylated in synucleinopathy lesions. Nature Cell Biology, 2, 160-164.
- Sevcsik, E., Trexler, A.J., Dunn, J.M. and Rhoades, E. (2011) Allostery in a disordered protein: Oxidative modifications to alpha-synuclein act distally to regulate membrane binding. Journal of the American Chemical Society, 18, 7152-7158. doi:10.1021/ja2009554
- Somogyi, A., Rosta, K., Pusztai, P., Tulassay, Z. and Nagy, G. (2007) Antioxidant measurements. Physiological Measurement, 4, R41-55. doi:10.1088/0967-3334/28/4/R01
- Segura-Aguilar, J. and Lind, C. (1989) On the mechanism of the Mn3(+)-induced neurotoxicity of dopamine: Prevention of quinone-derived oxygen toxicity by DT diaphorase and superoxide dismutase. Chemico-Biological Interactions, 3, 309-324. doi:10.1016/0009-2797(89)90006-9
- Napolitano, A., Manini, P. and d’Ischia, M. (2011) Oxidation chemistry of catecholamines and neuronal degeneration: An update. Current Medicinal Chemistry, 12, 1832- 1845. doi:10.2174/092986711795496863
- Gerlach, M., Double, K.L., Ben-Shachar, D., Zecca, L., Youdim, M.B. and Riederer, P. (2003) Neuromelanin and its interaction with iron as a potential risk factor for dopaminergic neurodegeneration underlying Parkinson’s disease. Neurotoxicity Research, 1-2, 35-44. doi:10.1007/BF03033371
- Fasano, M., Bergamasco, B. and Lopiano, L. (2006) Modifications of the iron-neuromelanin system in Parkinson’s disease. Journal of Neurochemistry, 4, 909-916. doi:10.1111/j.1471-4159.2005.03638.x
- Wolozin, B. and Golts, N. (2002) Iron and Parkinson’s disease. Neuroscientist, 1, 22-32. doi:10.1177/107385840200800107
- Zecca, L., Tampellini, D., Gerlach, M., Riederer, P., Fariello, R.G. and Sulzer, D. (2001) Substantia nigra neuromelanin: structure, synthesis, and molecular behaviour. Molecular Pathology, 6, 414-418.
- Ostrerova-Golts, N., Petrucelli, L., Hardy, J., Lee, J.M., Farer, M. and Wolozin, B. (2000) The A53T alpha-synuclein mutation increases iron-dependent aggregation and toxicity. The Journal of Neuroscience, 16, 6048-6054.
- Turnbull, S., Tabner, B.J., El-Agnaf, O.M., Moore, S., Davies, Y. and Allsop, D. (2001) alpha-Synuclein implicated in Parkinson’s disease catalyses the formation of hydrogen peroxide in vitro. Free Radical Biology & Medicine, 10, 1163-1170. doi:10.1016/S0891-5849(01)00513-5
- Shamoto-Nagai, M., Maruyama, W., Yi, H., Akao, Y., Tribl, F., Gerlach, M., Osawa, T., Riederer, P. and Naoi, M. (2006) Neuromelanin induces oxidative stress in mitochondria through release of iron: Mechanism behind the inhibition of 26S proteasome. Journal of Neural Transmission, 5, 633-644. doi:10.1007/s00702-005-0410-5
- Braak, H., Ghebremedhin, E., Rub, U., Bratzke, H. and Del Tredici, K. (2004) Stages in the development of Parkinson’s disease-related pathology. Cell and Tissue Research, 1, 121-134. doi:10.1007/s00441-004-0956-9
- Uchiyama, M., Isse, K., Tanaka, K., Yokota, N., Hamamoto, M., Aida, S., Ito, Y., Yoshimura, M. and Okawa, M. (1995) Incidental Lewy body disease in a patient with REM sleep behavior disorder. Neurology, 4, 709-712. doi:10.1212/WNL.45.4.709
- Turner, R.S., D’Amato, C.J., Chervin, R.D. and Blaivas, M. (2000) The pathology of REM sleep behavior disorder with comorbid Lewy body dementia. Neurology, 11, 1730- 1732. doi:10.1212/WNL.55.11.1730
- Boeve, B.F., Silber, M.H., Saper, C.B., Ferman, T.J., Dickson, D.W., Parisi, J.E., Benarroch, E.E., Ahlskog, J.E., Smith, G.E., Caselli, R.C., Tippman-Peikert, M., Olson, E.J., Lin, S.C., Young, T., Wszolek, Z., Schenck, C. H., Mahowald, M.W., Castillo, P.R., Del Tredici, K. and Braak, H. (2007) Pathophysiology of REM sleep behaviiour disorder and relevance to neurodegenerative disease. Brain, Pt 11, 2770-2788. doi:10.1093/brain/awm056
- Alexander, G.E., DeLong, M.R. and Strick, P.L. (1986) Parallel organization of functionally segregated circuits linking basal ganglia and cortex. Annual Review of Neuroscience, 9, 357-381. doi:10.1146/annurev.ne.09.030186.002041
- Albin, R.L., Young, A.B. and Penney, J.B. (1989) The functional anatomy of basal ganglia disorders. Trends in Neurosciences, 10, 366-375. doi:10.1016/0166-2236(89)90074-X
- DeLong, M.R. (1990) Primate models of movement disorders of basal ganglia origin. Trends in Neurosciences, 7, 281-285. doi:10.1016/0166-2236(90)90110-V
- Bamford, N.S., Robinson, S., Palmiter, R.D., Joyce, J.A., Moore, C. and Meshul, C.K. (2004) Dopamine modulates release from corticostriatal terminals. The Journal of Neuroscience, 43, 9541-9552. doi:10.1523/JNEUROSCI.2891-04.2004
- Gerfen, C.R., Engber, T.M., Mahan, L.C., Susel, Z., Chase, T.N., Monsma, F.J., Jr. and Sibley, D.R. (1990) D1 and D2 dopamine receptor-regulated gene expression of striatonigral and striatopallidal neurons. Science, 4986, 1429-1432. doi:10.1126/science.2147780
- Kravitz, A.V., Freeze, B.S., Parker, P.R., Kay, K., Thwin, M.T., Deisseroth, K. and Kreitzer, A.C. (2010) Regulation of parkinsonian motor behaviours by optogenetic control of basal ganglia circuitry. Nature, 7306, 622-626. doi:10.1038/nature09159
- Kempster, P.A., O’Sullivan, S.S., Holton, J.L., Revesz, T. and Lees, A.J. (2010) Relationships between age and late progression of Parkinson’s disease: A clinico-pathological study. Brain, Pt 6, 1755-1762. doi:10.1093/brain/awq059
- Scarpa, M., Rigo, A., Maiorino, M., Ursini, F. and Gregolin, C. (1984) Formation of alpha-tocopherol radical and recycling of alpha-tocopherol by ascorbate during peroxidation of phosphatidylcholine liposomes. An electron paramagnetic resonance study. Biochimica et Biophysica Acta, 2, 215-219. doi:10.1016/0304-4165(84)90070-9
- Lew, M.F. (2011) The evidence for disease modification in Parkinson’s disease. International Journal of Neuroscience, 121, 18-26. doi:10.3109/00207454.2011.620194
- Fahn, S. (1991) An open trial of high-dosage antioxidants in early Parkinson’s disease. The American Journal of Clinical Nutrition, 1, 380S-382S.
- Burne, T.H., McGrath, J.J., Eyles, D.W. and Mackay-Sim, A. (2005) Behavioural characterization of vitamin D receptor knockout mice. Behavioural Brain Research, 2, 299-308. doi:10.1016/j.bbr.2004.07.008
- Baluchnejadmojarad, T., Roghani, M., Nadoushan, M.R. and Bagheri, M. (2009) Neuroprotective effect of genistein in 6-hydroxydopamine hemi-parkinsonian rat model. Phytotherapy Research, 1, 132-135. doi:10.1002/ptr.2564
- [61] Bousquet, M., Calon, F. and Cicchetti, F. (2011) Impact of omega-3 fatty acids in Parkinson's disease. Ageing Research Reviews, 4, 453-463. doi:10.1016/j.arr.2011.03.001
- [62] Chang, Y.L., Chen, S.J., Kao, C.L., Hung, S.C., Ding, D.C., Yu, C.C., Chen, Y.J., Ku, H.H., Lin, C.P., Lee, K.H. Y., Chen, C., Wang, J.J., Hsu, C.C., Chen, L.K., Li, H.Y. and Chiou, S.H. (2012) Docosahexaenoic acid promotes dopaminergic differentiation in induced pluripotent stem cells and inhibits teratoma formation in rats with Parkinson-like pathology. Cell Transplantation, 1, 313-332.
- [63] Sakayori, N., Maekawa, M., Numayama-Tsuruta, K., Katura, T., Moriya, T. and Osumi, N. (2011) Distinctive effects of arachidonic acid and docosahexaenoic acid on neural stem /progenitor cells. Genes Cells, 7, 778-790. doi:10.1111/j.1365-2443.2011.01527.x
- [64] Li, S.C., Schoenberg, B.S., Wang, C.C., Cheng, X.M., Rui, D.Y., Bolis, C.L. and Schoenberg, D.G. (1985) A prevalence survey of Parkinson’s disease and other movement disorders in the People’s Republic of China. Archives of Neurology, 7, 655-657. doi:10.1001/archneur.1985.04060070045013
- [65] Zhang, Z.X. and Roman, G.C. (1993) Worldwide occurrence of Parkinson’s disease: An updated review. Neuroepidemiology, 4, 195-208. doi:10.1159/000110318
- [66] Barranco Quintana, J.L., Allam, M.F., Del Castillo, A.S. and Navajas, R.F. (2009) Parkinson’s disease and tea: A quantitative review. Journal of the American College of Nutrition, 1, 1-6.
- [67] Zhang, X., Xie, W., Qu, S., Pan, T., Wang, X. and Le, W. (2005) Neuroprotection by iron chelator against proteasome inhibitor-induced nigral degeneration. Biochemical and Biophysical Research Communication, 2, 544-549. doi:10.1016/j.bbrc.2005.05.150
- [68] Gal, M., Hromadova, M., Pospisil, L., Hives, J., Sokolova, R., Kolivoska, V. and Bulickova, J. (2010) Voltammetry of hypoxic cells radiosensitizer etanidazole radical anion in water. Bioelectrochemistry, 2, 118-123. doi:10.1016/j.bioelechem.2009.08.008
- [69] Ghosh, B., Antonio, T., Reith, M.E. and Dutta, A.K. (2010) Discovery of 4-(4-(2-((5-Hydroxy-1,2,3,4-tetrahydronaphthalen-2-yl)(propyl)amino)ethyl)piperazin-1-yl) quinolin-8-ol and its analogues as highly potent dopamine D2/D3 agonists and as iron chelator: In vivo activity indicates potential application in symptomatic and neuroprotective therapy for Parkinson’s disease. Journal of the Medicinal Chemistry, 5, 2114-2125. doi:10.1021/jm901618d
- [70] Ascherio, A., Weisskopf, M.G., O’Reilly, E.J., McCullough, M.L., Calle, E.E., Rodriguez, C. and Thun, M.J. (2004) Coffee consumption, gender, and Parkinson’s disease mortality in the cancer prevention study II cohort: The modifying effects of estrogen. American Journal of Epidemiology, 10, 977-984. doi:10.1093/aje/kwh312
- [71] Fredholm, B.B., Battig, K., Holmen, J., Nehlig, A. and Zvartau, E.E. (1999) Actions of caffeine in the brain with special reference to factors that contribute to its widespread use. Pharmacological Reviews, 1, 83-133.
- [72] Trinh, K., Andrews, L., Krause, J., Hanak, T., Lee, D., Gelb, M. and Pallanck, L. (2010) Decaffeinated coffee and nicotine-free tobacco provide neuroprotection in Drosophila models of Parkinson’s disease through an NRF2-dependent mechanism. The Journal of Neuroscience, 16, 5525-5532. doi:10.1523/JNEUROSCI.4777-09.2010
- [73] Simoes, A.P., Duarte, J.A., Agasse, F., Canas, P.M., Tome, A.R., Agostinho, P. and Cunha, R.A. (2012) Blockade of adenosine A2A receptors prevents interleukin-1beta-induced exacerbation of neuronal toxicity through a p38 mitogen-activated protein kinase pathway. Journal of Neuroinflammation, 204. doi:10.1186/1742-2094-9-204
- [74] Alfinito, P.D., Wang, S.P., Manzino, L., Rijhsinghani, S., Zeevalk, G.D. and Sonsalla, P.K. (2003) Adenosinergic protection of dopaminergic and GABAergic neurons against mitochondrial inhibition through receptors located in the substantia nigra and striatum, respectively. The Journal of Neuroscience, 34, 10982-10987.
- [75] Birkmayer, W., Riederer, P., Youdim, M.B. and Linauer, W. (1975) The potentiation of the anti akinetic effect after L-dopa treatment by an inhibitor of MAO-B, Deprenil. Journal of Neural Transmission, 3-4, 303-326. doi:10.1007/BF01253131
- [76] Riederer, P. and Laux, G. (2011) MAO-inhibitors in Parkinson’s disease. Experimental Neurobiology, 1, 1-17.
- [77] Jenner, P. and Langston, J.W. (2011) Explaining ADAGIO: A critical review of the biological basis for the clinical effects of rasagiline. Movement Disorders, 13, 2316-2323. doi:10.1002/mds.23926
- [78] Nyholm, D., Johansson, A., Lennernas, H. and Askmark, H. (2012) Levodopa infusion combined with entacapone or tolcapone in Parkinson disease: A pilot trial. European Journal of Neurology, 6, 820-826. doi:10.1111/j.1468-1331.2011.03614.x
- [79] Hauser, R.A., Schapira, A.H., Rascol, O., Barone, P., Mizuno, Y., Salin, L., Haaksma, M., Juhel, N. and Poewe, W. (2010) Randomized, double-blind, multicenter evaluation of pramipexole extended release once daily in early Parkinson's disease. Movement Disorders, 15, 2542-2549. doi:10.1002/mds.23317
- [80] Whone, A.L., Watts, R.L., Stoessl, A.J., Davis, M., Reske, S., Nahmias, C., Lang, A.E., Rascol, O., Ribeiro, M.J., Remy, P., Poewe, W.H., Hauser, R.A. and Brooks, D.J. (2003) Slower progression of Parkinson’s disease with ropinirole versus levodopa: The REAL-PET study. Annals of Neurology, 1, 93-101. doi:10.1002/ana.10609
- [81] (2003) A controlled trial of rotigotine monotherapy in early Parkinson’s disease. Archives of Neurology, 12, 1721- 1728.
- [82] Jones, C.A., Johnston, L.C., Jackson, M.J., Smith, L.A., van Scharrenburg, G., Rose, S., Jenner, P.G. and McCreary, A.C. (2010) An in vivo pharmacological evaluation of pardoprunox (SLV308)—a novel combined dopamine D(2)/D(3) receptor partial agonist and 5-HT(1A) receptor agonist with efficacy in experimental models of Parkinson's disease. European Neuropsychopharmacology, 8, 582-593. doi:10.1016/j.euroneuro.2010.03.001
- [83] Rascol, O., Bronzova, J., Hauser, R.A., Lang, A.E., Sampaio, C., Theeuwes A. and van de Witte, S.V. (2012) Pardoprunox as adjunct therapy to levodopa in patients with Parkinson’s disease experiencing motor fluctuations: Results of a double-blind, randomized, placebo-controlled, trial. Parkinsonism & Related Disorders, 4, 370-376. doi:10.1016/j.parkreldis.2011.12.006
- [84] Bonuccelli, U., Piccini, P. and Rabey, J.M. (2009) Old and new dopamine agonists in Parkinson’s disease: A reappraisal. Introduction. Parkinsonism & Related Disorders, 15, S1. doi:10.1016/S1353-8020(09)00312-5
- [85] Yoshioka, M., Tanaka, K., Miyazaki, I., Fujita, N., Higashi, Y., Asanuma, M. and Ogawa, N. (2002) The dopamine agonist cabergoline provides neuroprotection by activation of the glutathione system and scavenging free radicals. Neuroscience Research, 3, 259-267. doi:10.1016/S0168-0102(02)00040-8
- [86] Katayama, S. (2001) Actigraph analysis of diurnal motor fluctuations during dopamine agonist therapy. European Neurology, 46, 11-17. doi:10.1159/000058048
- [87] Bracco, F., Battaglia, A., Chouza, C., Dupont, E., Gershanik, O., Marti Masso, J.F. and Montastruc, J.L. (2004) The long-acting dopamine receptor agonist cabergoline in early Parkinson’s disease: Final results of a 5-year, doubleblind, levodopa-controlled study. CNS Drugs, 11, 733- 746. doi:10.2165/00023210-200418110-00003
- [88] Calabresi, P., Centonze, D., Gubellini, P., Pisani, A. and Bernardi, G. (2000) Acetylcholine-mediated modulation of striatal function. Trends Neuroscience, 3, 120-126. doi:10.1016/S0166-2236(99)01501-5
- [89] Luginger, E., Wenning, G.K., Bosch, S. and Poewe, W. (2000) Beneficial effects of amantadine on L-dopa-induced dyskinesias in Parkinson’s disease. Movement Disorders, 5, 873-878. doi:10.1002/1531-8257(200009)15:5<873::AID-MDS1017>3.0.CO;2-I
- [90] Crosby, N.J., Deane, K.H. and Clarke, C.E. (2003) Amantadine for dyskinesia in Parkinson’s disease. The Cochrane Database of Systematic Reviews, 2, CD003467.
- [91] Gorell, J.M., Rybicki, B.A., Johnson, C.C. and Peterson, E.L. (1999) Smoking and Parkinson’s disease: A dose-response relationship. Neurology, 1, 115-119. doi:10.1212/WNL.52.1.115
- [92] Baron, J.A. (1986) Cigarette smoking and Parkinson’s disease. Neurology, 11, 1490-1496. doi:10.1212/WNL.36.11.1490
- [93] Grandinetti, A., Morens, D.M., Reed, D. and MacEachern, D. (1994) Prospective study of cigarette smoking and the risk of developing idiopathic Parkinson’s disease. American Journal of Epidemiology, 12, 1129-1138.
- [94] Morens, D.M., Grandinetti, A., Davis, J.W., Ross, G.W., White, L.R. and Reed, D. (1996) Evidence against the operation of selective mortality in explaining the association between cigarette smoking and reduced occurrence of idiopathic Parkinson disease. American Journal of Epidemiology, 4, 400-404. doi:10.1093/oxfordjournals.aje.a008941
- [95] Thacker, E.L., O’Reilly, E.J., Weisskopf, M.G., Chen, H., Schwarzschild, M.A., McCullough, M.L., Calle, E.E., Thun, M.J. and Ascherio, A. (2007) Temporal relationship between cigarette smoking and risk of Parkinson disease. Neurology, 10, 764-768. doi:10.1212/01.wnl.0000256374.50227.4b
- [96] Quik, M. (2004) Smoking, nicotine and Parkinson’s disease. Trends in Neuroscience, 9, 561-568. doi:10.1016/j.tins.2004.06.008
- [97] Jeyarasasingam, G., Tompkins, L. and Quik, M. (2002) Stimulation of non-alpha7 nicotinic receptors partially protects dopaminergic neurons from 1-methyl-4-phenylpyridinium-induced toxicity in culture. Neuroscience, 2, 275-285. doi:10.1016/S0306-4522(01)00488-2
- [98] Ryan, R.E., Ross, S.A., Drago, J. and Loiacono, R.E. (2001) Dose-related neuroprotective effects of chronic nicotine in 6-hydroxydopamine treated rats, and loss of neuroprotection in alpha4 nicotinic receptor subunit knockout mice. British Journal of Pharmacology, 8, 1650- 1656. doi:10.1038/sj.bjp.0703989
- [99] Soto-Otero, R., Mendez-Alvarez, E., Hermida-Ameijeiras, A., Lopez-Real, A.M. and Labandeira-Garcia, J.L. (2002) Effects of (-)-nicotine and (-)-cotinine on 6-hydroxy-dopamine-induced oxidative stress and neurotoxicity: Relevance for Parkinson’s disease. Biochemical Pharmacology, 1, 125-135. doi:10.1016/S0006-2952(02)01070-5
- [100] Visanji, N.P., O’Neill, M.J. and Duty, S. (2006) Nicotine, but neither the alpha4beta2 ligand RJR2403 nor an alpha7 nAChR subtype selective agonist, protects against a partial 6-hydroxydopamine lesion of the rat median forebrain bundle. Neuropharmacology, 3, 506-516. doi:10.1016/j.neuropharm.2006.04.015
- [101] Quik, M., Parameswaran, N., McCallum, S.E., Bordia, T., Bao, S., McCormack, A., Kim, A., Tyndale, R.F., Langston, J.W. and Di Monte, D.A. (2006) Chronic oral nicotine treatment protects against striatal degeneration in MPTPtreated primates. Journal of Neurochemistry, 6, 1866- 1875. doi:10.1111/j.1471-4159.2006.04078.x
- [102] Bordia, T., Parameswaran, N., Fan, H., Langston, J.W., McIntosh, J.M. and Quik, M. (2006) Partial recovery of striatal nicotinic receptors in 1-methyl-4-phenyl-1,2,3,6- tetrahydropyridine (MPTP)-lesioned monkeys with chronic oral nicotine. Journal of Pharmacology and Experimental Therapeutics, 1, 285-292. doi:10.1124/jpet.106.106997
- [103] Costa, G., Abin-Carriquiry, J.A. and Dajas, F. (2001) Nicotine prevents striatal dopamine loss produced by 6-hydroxydopamine lesion in the substantia nigra. Brain Research, 2, 336-342. doi:10.1016/S0006-8993(00)03087-0
- [104] Behmand, R.A. and Harik, S.I. (1992) Nicotine enhances 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine neurotoxicity. Journal of Neurochemistry, 2, 776-779. doi:10.1111/j.1471-4159.1992.tb09786.x
- [105] Moll, H. (1926) The treatment of post-encephalitig parkinsonism by nicotine. British Medical Journal, 3416, 1079-1081. doi:10.1136/bmj.1.3416.1079
- [106] Villafane, G., Cesaro, P., Rialland, A., Baloul, S., Azimi, S., Bourdet, C., Le Houezec, J., Macquin-Mavier, I. and Maison, P. (2007) Chronic high dose transdermal nicotine in Parkinson’s disease: An open trial. European Journal of Neurology, 12, 1313-1316. doi:10.1111/j.1468-1331.2007.01949.x
- [107] Fagerstrom, K.O., Pomerleau, O., Giordani, B. and Stelson, F. (1994) Nicotine may relieve symptoms of Parkinson’s disease. Psychopharmacology (Berl), 1, 117-119. doi:10.1007/BF02244882
- [108] Bordia, T., Campos, C., Huang, L. and Quik, M. (2008) Continuous and intermittent nicotine treatment reduces L-3,4-dihydroxyphenylalanine (L-DOPA)-induced dyskinesias in a rat model of Parkinson’s disease. Journal of Pharmacology and Experimental Therapeutics, 1, 239- 247. doi:10.1124/jpet.108.140897
- [109] Picciotto, M.R. and Zoli, M. (2008) Neuroprotection via nAChRs: The role of nAChRs in neurodegenerative disorders such as Alzheimer’s and Parkinson’s disease. Frontiers in Bioscience, 13, 492-504. doi:10.2741/2695
- [110] Lindvall, O., Bjorklund, A. and Skagerberg, G. (1984) Selective histochemical demonstration of dopamine terminal systems in rat diand telencephalon: new evidence for dopaminergic innervation of hypothalamic neurosecretory nuclei. Brain Research, 1-2, 19-30. doi:10.1016/0006-8993(84)90352-4
- [111] Dehorter, N., Guigoni, C., Lopez, C., Hirsch, J., Eusebio, A., Ben-Ari, Y. and Hammond, C. (2009) Dopamine-deprived striatal GABAergic interneurons burst and generate repetitive gigantic IPSCs in medium spiny neurons. The Journal of Neuroscience, 24, 7776-7787. doi:10.1523/JNEUROSCI.1527-09.2009
- [112] Xiao, C., Nashmi, R., McKinney, S., Cai, H., McIntosh, J. M. and Lester, H.A. (2009) Chronic nicotine selectively enhances alpha4beta2* nicotinic acetylcholine receptors in the nigrostriatal dopamine pathway. The Journal of Neuroscience, 40, 12428-12439. doi:10.1523/JNEUROSCI.2939-09.2009
- [113] Gittis, A.H., Leventhal, D.K., Fensterheim, B.A., Pettibone, J.R., Berke, J.D. and Kreitzer, A.C. (2011) Selective inhibition of striatal fast-spiking interneurons causes dyskinesias. The Journal of Neuroscience, 44, 15727- 15731. doi:10.1523/JNEUROSCI.3875-11.2011
- [114] Gregorio, M.L., Wietzikoski, E.C., Ferro, M.M., Silveira, J.L., Vital, M.A. and Da Cunha, C. (2009) Nicotine induces sensitization of turning behavior in 6-hydroxy-dopamine lesioned rats. Neurotoxicity Research, 4, 359-366. doi:10.1007/s12640-009-9041-1
- [115] Zhao-Shea, R., Cohen, B.N., Just, H., McClure-Begley, T., Whiteaker, P., Grady, S.R., Salminen, O., Gardner, P.D., Lester, H.A. and Tapper, A.R. (2010) Dopamine D2-receptor activation elicits akinesia, rigidity, catalepsy, and tremor in mice expressing hypersensitive {alpha}4 nicotinic receptors via a cholinergic-dependent mechanism. The FASEB Journal, 1, 49-57. doi:10.1096/fj.09-137034
- [116] Kalivas, P.W., Churchill, L. and Klitenick, M.A. (1993) GABA and enkephalin projection from the nucleus accumbens and ventral pallidum to the ventral tegmental area. Neuroscience, 4, 1047-1060. doi:10.1016/0306-4522(93)90048-K
- [117] McGehee, D.S., Heath, M.J., Gelber, S., Devay, P. and Role, L.W. (1995) Nicotine enhancement of fast excitatory synaptic transmission in CNS by presynaptic receptors. Science, 5231, 1692-1696. doi:10.1126/science.7569895
- [118] Ribeiro, E.B., Bettiker, R.L., Bogdanov, M. and Wurtman, R.J. (1993) Effects of systemic nicotine on serotonin release in rat brain. Brain Research, 2, 311-318. doi:10.1016/0006-8993(93)90121-3
- [119] Boyadjieva, N.I. and Sarkar, D.K. (1997) The secretory response of hypothalamic beta-endorphin neurons to acute and chronic nicotine treatments and following nicotine withdrawal. Life Science, 6, PL59-66.
- [120] Pierzchala, K., Houdi, A.A. and Van Loon, G.R. (1987) Nicotine-induced alterations in brain regional concentrations of native and cryptic Metand Leu-enkephalin. Peptides, 6, 1035-1043. doi:10.1016/0196-9781(87)90133-1
- [121] de Rover, M., Lodder, J.C., Kits, K.S., Schoffelmeer, A.N. and Brussaard, A.B. (2002) Cholinergic modulation of nucleus accumbens medium spiny neurons. European Journal of Neuroscience, 12, 2279-2290. doi:10.1046/j.1460-9568.2002.02289.x
- [122] Gittis, A.H., Nelson, A.B., Thwin, M.T., Palop, J.J. and Kreitzer, A.C. (2010) Distinct roles of GABAergic interneurons in the regulation of striatal output pathways. The Journal of Neuroscience, 6, 2223-2234. doi:10.1523/JNEUROSCI.4870-09.2010
- [123] García-Montes, J.R., Boronat-García, A., López-Colomé, A.M., Bargas, J., Guerra-Crespo, M. and Drucker-Colín, R. (2012) Is nicotine protective against Parkinson’s disease? An experimental analysis. CNS & Neurological Disorders-Drug Targets, in press.