Materials Sciences and Applications
Vol.4 No.6(2013), Article ID:33033,9 pages DOI:10.4236/msa.2013.46047
Gelation of Antheraea pernyi Silk Fibroin Accelerated by Shearing
![]()
1National Engineering Laboratory for Modern Silk, College of Textile and Clothing Engineering, Soochow University, Suzhou, China; 2Suzhou Institute of Scientific & Technical Information, Suzhou, China.
Email: *mzli@suda.edu.cn
Copyright © 2013 Yu Liu et al. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Received April 16th, 2013; revised May 15th, 2013; accepted May 24th, 2013
Keywords: Silk Fibroin; Gels; Shearing; Structural Transition
ABSTRACT
The rapid manufacture of silk fibroin gels in mild conditions is an important subject in the field of silk-based biomaterials. In this study, the gelation of Antheraea pernyi silk fibroin (ASF) aqueous solution was induced by shearing, without chemical cross-linking agents. Simple shearing controlled and accomplished the steady and rapid conformational transition to β-sheets with ease. The conformational transformation and rapid gelation mechanisms of ASF induced by shearing were tracked and analyzed by circular dichroism spectrometry, Fourier transform infrared spectroscopy and X-ray diffractometry, then compared with Bombyx mori silk fibroin (BSF). ASF quickly formed hydrogels within 24 - 48 h after shearing under different shearing rates for 30 - 90 min, resulting in sol-gel transformation when the β-sheet content reached nearly 50%, which is the minimum content needed to maintain a stable hydrogel system in ASF. The gel structures remained stable once formed. The rapid gelation of ASF through shearing compared with BSF was achieved because of ASF’s alternating polyalanine-containing units, which tend to form α-helix structures spontaneously. Further, the entropic cost during the conformational transition from the α-helix to the β-sheet structure is less than the cost of the transition from the random coil structure. This method is a simple, non-chemical cross-linking approach for the promotion of rapid gelation and the protection of the biological properties of ASF, and it may prove useful for application in the field of biomedical materials.
1. Introduction
Silk fibroin (SF) is a natural structural protein excreted from domestic (Bombyx mori) or wild silkworms. The former has been widely used in the textile industry for thousands of years due to its outstanding mechanical properties [1]. In recent years, the impressive biocompatibility and biodegradation properties [2,3] of Bombyx mori silk fibroin (BSF) have made it a popular starting material for tissue engineering scaffolds, such as blood [4], skin [5], bone [6], ligament [7], nerve [8] and cornea [9], and for controlled drug release delivery [10]. As one of the most common species of wild silkworms, Antheraea pernyi silk fibroin (ASF) is characterized by more Ala, Arg and His and fewer Gly amino acid residues than BSF. It also contains the tripeptide sequence Arg-GlyAsp (RGD), which is known to be the receptor of cell integrins and to mediate special interactions between mammalian cells [11,12]. Based on these features, interest has arisen in the use of ASF as an advanced biomedical application material in such fields as cell culture substrates and tissue engineering scaffolds [13-15]. For example, Patra et al. [16] evaluated the potential application of A. mylitta silk fibroin as a biomaterial for threedimensional (3D) cardiac tissue engineering scaffolds. The results showed that A. mylitta silk fibroin was superior to BSF due to its RGD.
Hydrogels, both synthetic and natural, are a class of highly hydrated polymeric materials [17]. They can maintain a distinct 3D porous structure with mechanical and structural properties similar to the properties of many natural tissues and extra-cellular matrices [18]. As an important biomaterial form, SF hydrogels have been of interest for tissue engineering [19] and drug/growth factor delivery [20]. It is generally believed that SF in aqueous solution undergoes self-assembly into β-sheet structures and forms hydrogels in vitro. However, very long time may be required for the spontaneous gelation of SF, which limits its clinical application. SF hydrogels can also be produced by physical stimulants through hydrophobic/hydrophilic interactions [21], such as low pH, high temperature, high ionic strength, ultrasonication or vortex in vitro [22-25], but these harsh conditions may alter cell function and affect cell viability [26]. Several chemical additives have also been used to shorten the gelation time of SF, but the resultant hydrogels may suffer from the inherent toxicity of the cross-linking additives [27-29]. It has been reported that shearing was significantly more powerful than ultrasonic shaking for promoting the conformational transition from the random coil to the β-sheet of the BSF aqueous solution [30]. That is, a steady and rapid conformational transition can be easily accomplished by simple shearing.
ASF could be used in tissue engineering scaffolds and bioactive controlled release carriers if the sol-gel transformation could be completed rapidly under mild conditions. We have previously reported that the gelation time of ASF decreased with increasing ASF concentration and temperature [31], accompanied by an observable conformational transformation to β-sheets during the process of gelation. In this work, a simple physical method for promoting ASF gelation was developed by shearing under mild conditions, avoiding the complications and inherent toxicity to cells or cytokines caused by unpurified or remaining cross-linking additives, with emphasis on the mechanism by which shearing accelerates the molecular conformational transition and induces rapid ASF gelation.
2. Materials and Methods
2.1. Preparation of the SF Solutions
As described previously [15], A. pernyi silk purchased from Liaoning Province (China) was treated three times with 2.5 g/L Na2CO3 solution at 98˚C - 100˚C for 30 min to remove sericin. The degummed ASF fibers were dissolved in melted Ca (NO3)2·4H2O solutions with a 1:10 (w/v) bath ratio for 5 h at 105˚C. Then, the mixed solution was dialyzed against distilled water for 96 h at room temperature with a cellulose tube (WMCO 9000, Pierce) to remove the excess salts.
As previously described [3], B. mori fibers were degummed in a solution containing 0.6 g/L Na2CO3 at 98˚C - 100˚C for 30 min. This step was repeated three times, and the degummed fibers were dissolved in the triadic solvent CaCl2·CH3CH2OH·H2O (molar ratio = 1:2:8) at 72˚C ± 2˚C for 1 h with stirring. The BSF solution was obtained by dialyzing for 96 h at room temperature with a cellulose tube (WMCO 9000, Pierce) to remove the excess salts.
2.2. Preparation of the SF Gels
4 ml of ASF solution, at a concentration of 3.0 wt%, was sheared at defined rates (280, 380 and 480 rpm) for different times (15, 30, 45, 60 and 90 min) at 20˚C ± 2˚C using a JB50-D electric mixer (Shanghai specimen and model factory, China). Then, the solution was poured into culture dishes (a diameter of 5 cm), which were incubated in a Synergy HT Multi-Mode Microplate Reader (Bio-Tek Instruments, USA) at 20˚C ± 3˚C, and the gelation time of each sample was recorded. The gelation time was determined when the sample had developed an opaque white color and did not fall from an inverted dish within 30 s. The BSF gel preparation and the gelation time determination were performed in the same way.
2.3. Circular Dichroism (CD) Spectra
Measurements were taken using a J-715 spectropolarimeter equipped with a slab (NESLAB RTE-111) and purged with N2 gas at a flow rate of 3 - 5 mL/min. The SF aqueous solution was introduced into a cell with a 0.1 cm path length for far-UV measurement at a protein concentration of 0.1 mg/mL. CD spectra were recorded from 190 to 250 nm wavelengths with a resolution of 0.2 nm and an accumulation of five scans at a scanning rate of 100 nm/min and a response time of 0.25 s. The scans were accumulated and averaged for each spectrum after subtracting the background of blank water. The conformational contents of the SF spectra were determined using the CD spectra deconvolution program (CDNN 2.1 software).
2.4. X-Ray Diffraction (XRD) Spectra
The freeze-dried gels were cut into micro-powders with diameters less than 40 µm and measured by wide-angle X-ray diffraction (XRD). The diffraction intensities were measured using a Rigaku D/Max-3C diffractometer with Cu-Kα radiation (λ = 0.15406 nm) at the scanning rate of 2˚/min and a range of 2θ from 5˚ to 45˚. The X-ray source was operated at 40 kV and 40 mA.
2.5. Fourier Transform Infrared (FTIR) Spectra
The freeze-dried gels were cut into micro-particles with diameters less than 40 µm, from which the samples were prepared into KBr pellets. Fourier transform infrared (FTIR) spectra were obtained using a Nicolet Avatar-IR 360 in the spectral region of 400 - 4000 cm−1.
3. Results and Discussion
3.1. The Gelation Time of SF
The relationship between SF gelation time and shearing rate/time at 20˚C ± 2˚C is shown in Figure 1.
With increasing shearing rate and time, the gelation time of ASF decrease sharply (Figure 1(a)). The sol-gel transformation of the ASF solution was completed within 24 - 48 h by shearing for 30 - 90 min at different rates (280, 380, 480 rpm). As shown in Figure 1(b), the gelation time of BSF was not significantly decreased initially (0 - 30 min) but did decrease with increasing shearing time from 30 to 90 min. The results show that the gelation rate of ASF was significantly faster than the gelation rate of BSF under the same conditions. Additionally, the gelation behaviors of ASF and BSF were significantly distinct. The shearing influenced the gelation rate of ASF more than that of BSF and promoted the completion of the ASF sol-gel transition within 1 - 2 days.
3.2. The Molecular Conformation of SF
CD is generally used to test different protein conformations in dilute aqueous solutions. Studies have shown that the secondary structure (α-helix, β-sheet, β-turn and random coil) of a protein exhibits characteristic peaks in the far-UV zone (178 - 250 nm). For example, a strong negative peak at 216 nm and a strong positive peak
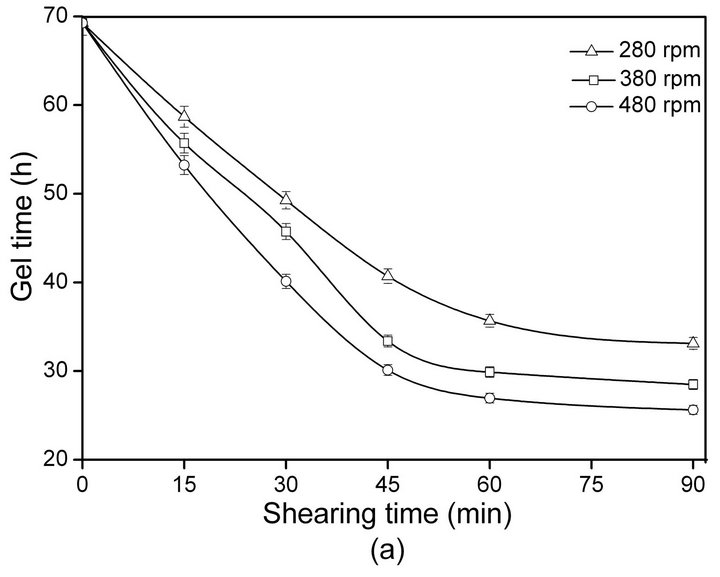
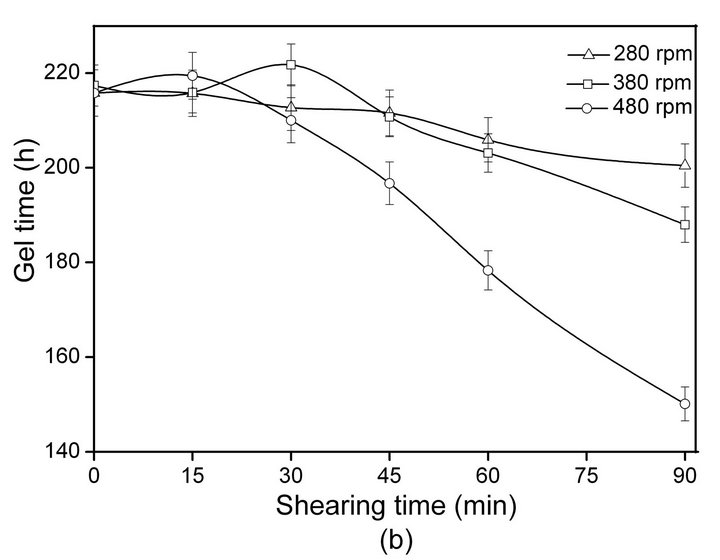
Figure 1. The influence of shearing time and rate on the gelation time of SF: ASF (a), BSF (b).
(positive cotton effect) at 185 - 200 nm are the characteristic peaks of the β-sheet structure, and peaks at 192 nm (positive), 208 nm (negative) and 222 nm (negative) are characteristic of the α-helix structure, while a strong negative peak (negative cotton effect) at approximately 195 nm is characteristic of the random coil [32,33].
To confirm the secondary structure changes in the shearing-induced SF gels, CD spectra were scanned at a defined shearing rate and time (Figures 2 and 3). As shown in Figure 2 curve a, the initial ASF solution without shearing showed strong peaks at approximately 206 nm (negative) and 217 nm (negative) and a weak peak at approximately 220 nm (negative), indicating that the molecular conformation of ASF was initially composed predominantly of α-helix with some β-sheet structures. The conformation of ASF gradually changed with increasing shearing rate. The negative peak at 206 nm became weaker with the simultaneous increase of the negative peak at 217 nm, demonstrating the formation of β-sheets during shearing (Figure 2, curves b, c, d). Shearing initiated the formation of β-sheets and accelerated the formation of the physical cross-links responsible for gels stabilization. With increasing shearing rate, the ASF molecules along the shearing direction changed to a well-ordered arrangement, forming abundant hydrogen bonds and enhancing the hydrophobic interaction. Therefore, the high shearing rate accelerated the molecular movement, promoting the rapid formation of β-sheets.
The transformation of the random-coil structure of BSF molecules into the β-sheet structure is shown in Figure 3. The initial BSF solution without shearing displayed a strong negative peak at approximately 196 nm but no distinct peaks near 217 and 209 nm (curve a),
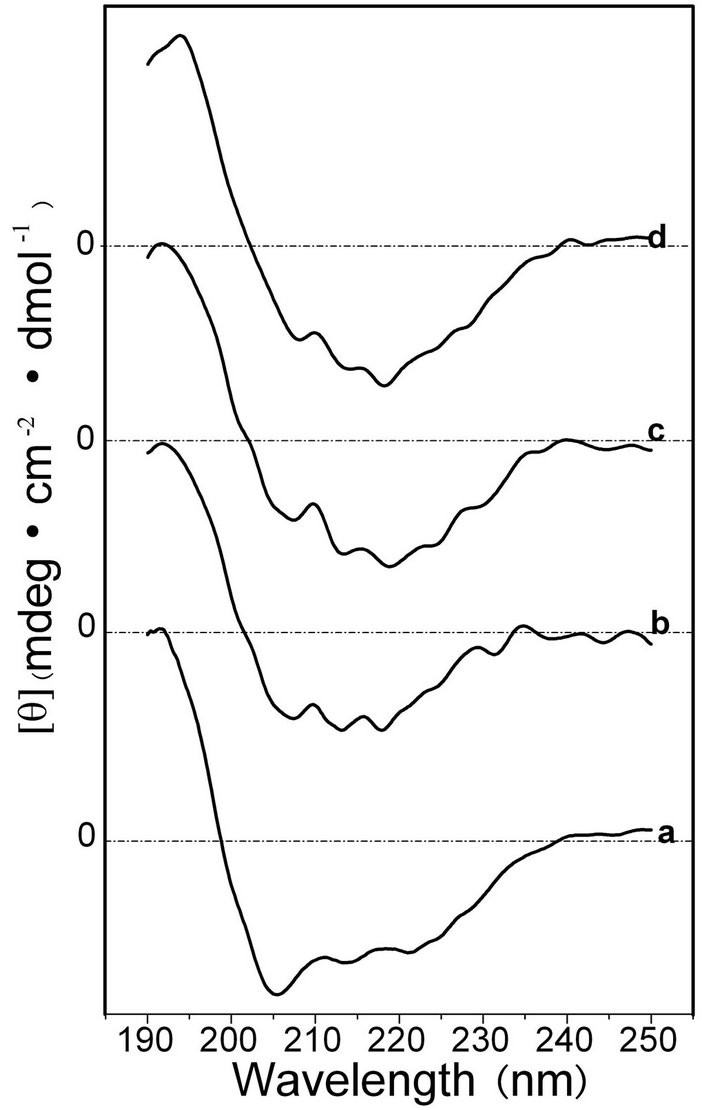
Figure 2. CD spectra of the ASF solution after shearing for 90 min at shearing rates of 0 rpm (a), 280 rpm, (b) 380 rpm (c) and 480 rpm (d).
showing the typical random coil conformation. With increasing shearing rate, the negative peaks at 217 nm developed and intensified, illustrating that shearing promoted the conformational transition of BSF from random coil to β-sheet (curves b, c and d).
The percentages of molecular conformations in SF solutions after shearing for 90 min at various rates were calculated based on the experimentally obtained curves, as shown in Table 1. Significant differences were identified in the molecular conformations between the initial ASF and BSF solutions. The contents of the α-helix and β-sheet were 42.0% and 12.1% in the initial ASF solution, respectively, but were not detected in the initial BSF solution. In contrast, the initial BSF solution contained up to 80.1% random coil content, which was more than in ASF (45.9%). Moreover, the molecular conformation contents of both the ASF and BSF solutions quickly changed during the shearing process. After shearing for 90 min at 480 rpm, a pronounced growth in the β-sheet content of ASF (from 12.1% to 37.5%) was observed, accompanied by a decrease in the random coil and α-helix contents (from 45.9% to 21.8% and from 42.0% to 32.8%, respectively). Correspondingly, the random coil content of BSF declined from 80.1% to 48.8%, and the β-sheet content increased to 29.6%. This result illustrates that the β-sheet fraction formed more easily in ASF than in BSF with increasing shearing rate.
After shearing for 90 min at 480 rpm, the SF solutions were incubated for 24 h at 20˚C, and then the corresponding secondary structure contents of SF were measured, as shown in Table 2. After incubating for 24 h, the molecular conformations of both the ASF and the BSF solutions changed significantly. The α-helix and random coil contents of ASF decreased from 32.8% to 18.2%, and 21.8% to 5.0%, respectively, and the β-sheet content increased by 12.5% (from 37.5% to 50.0%). The random coil content of BSF declined from 48.8% to 33.8%, and the β-sheet content increased by 8.5% (from 29.6% to 38.1%) under the same conditions. The increase in the rate of β-sheet content formation in the ASF solution was greater than in BSF during the process of incubation after shearing. The results indicate that the α-helix structure in the ASF initial solution was easier to transform into β-sheets than the random coil structure in the BSF solution.
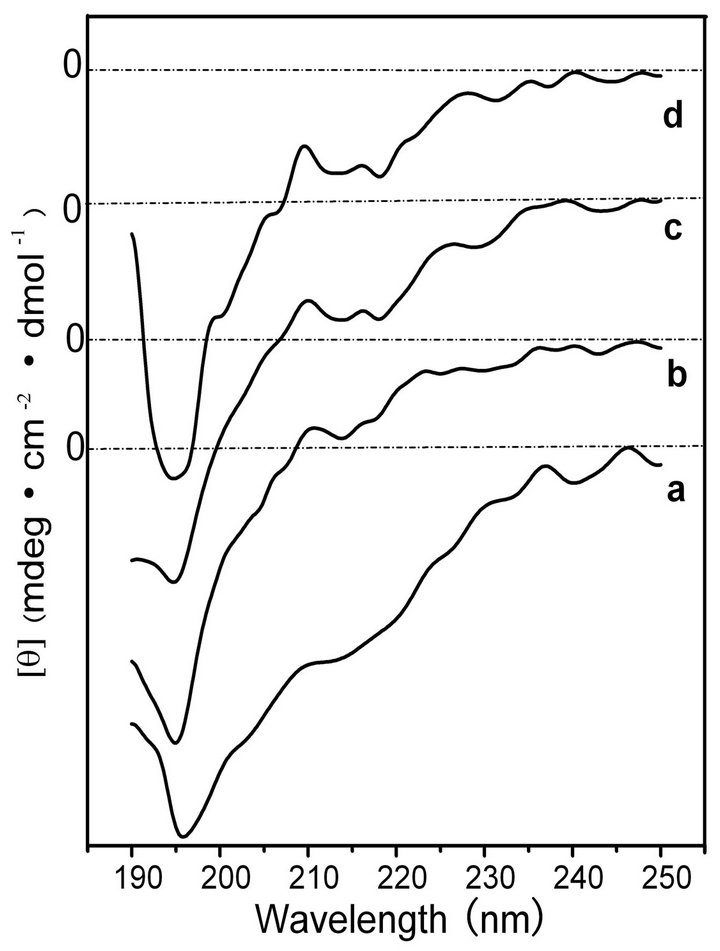
Figure 3. CD spectra of the BSF solution after shearing for 90 min at shearing rates of 0 rpm (a), 280 rpm, (b) 380 rpm (c) and 480 rpm (d).
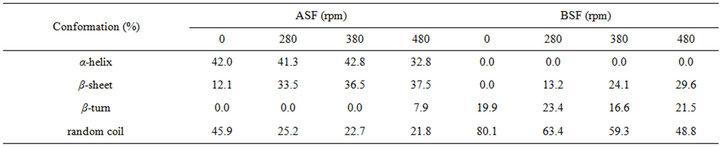
Table 1. The conformation contents for SF after shearing for 90 min at different rates.
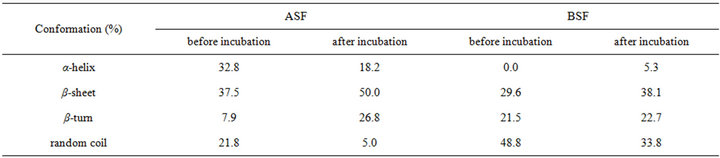
Table 2. The conformation contents of SF incubate for 24 h after shearing at 480 rpm for 90 min.
Comparing the results in Table 2 to Figure 1(a), an interesting phenomenon can be identified: after shearing for 90 min at 480 rpm and incubation for 24 h, the β- sheet content of ASF reached nearly 50% (Table 2), accompanied by the sol-gel transformation (Figure 1(a)). Thus, shearing provided a useful new tool to initiate the rapid sol-gel transitions of ASF, and the β-sheet content of 50% was the minimum content required to maintain a stable hydrogel system in ASF.
3.3. Structure of the SF Gels
Following previous studies on the molecular conformation of SF [34,35], the X-ray diffraction patterns contained peaks at 11.8˚ and 22.0˚ for the α-helix structure, and at 16.5˚, 20.2˚, 24.9˚, 30.90˚, 34.59˚, 40.97˚ and 44.12˚ for the β-sheet structure. Figures 4 and 5 show the X-ray profiles of freeze-dried gels prepared from the SF solutions treated by shearing at different rates. These patterns exhibit strong diffraction peaks at approximately 16.5˚, 20.2˚, and 24.9˚, indicating the presence of the β- sheet structure in the sample of ASF gels without shearing (shown in Figure 4 curve a). Additionally, the molecular conformations of the ASF gels processed with shearing (curves b, c, d) or without (curve a) contain no significant differences and were all mainly composed of β-sheets. In Figure 5 curve a, the peaks appeared at approximately 11.8˚, 20.7˚ and 24.7˚, indicating the presence of a typical β-sheet structure, with these results approximately in agreement with previous reports [15,36]. Moreover, with increasing shearing rate, these diffraction peaks (curves b, c and d) remained almost unchanged, illustrating that the gels consisted mainly of the β-sheet structure.
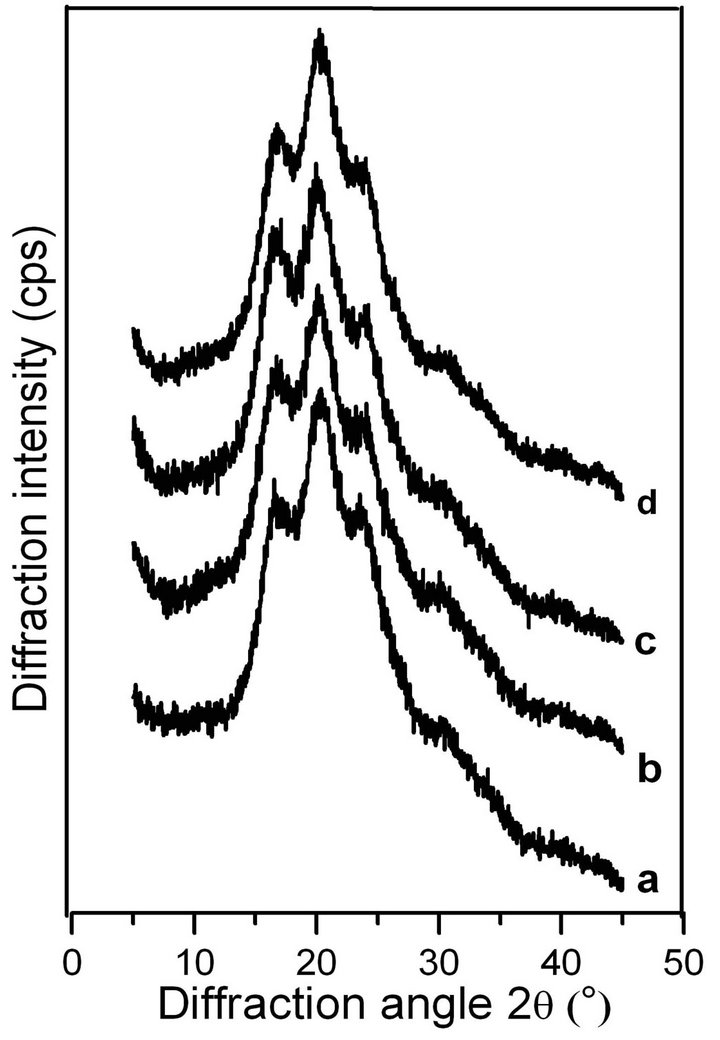
Figure 4. X-ray diffraction curves of the ASF gels induced by shearing for 90 min at rates of 0 rpm (a), 280 rpm (b), 380 rpm (c) and 480 rpm (d).
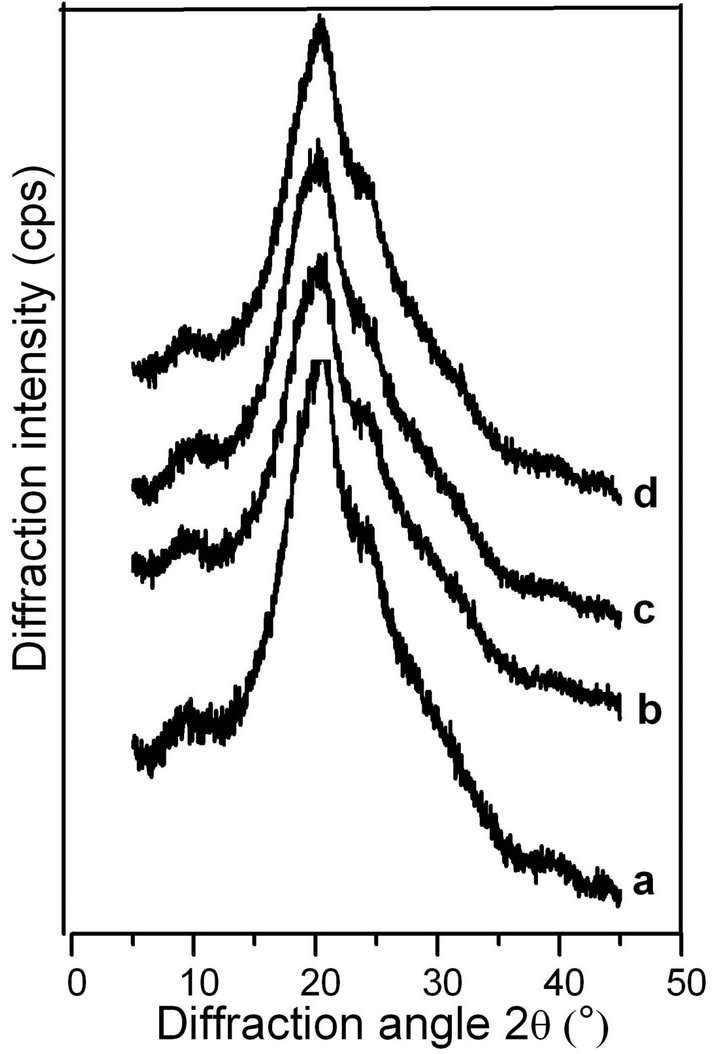
Figure 5. X-ray diffraction curves of the BSF gels induced by shearing for 90 min at rates of 0 rpm (a), 280 rpm (b), 380 rpm (c) and 480 rpm (d).
From Figures 4 and 5, it can be concluded that the shearing induced the gelation of SF by promoting the conformational transition from α-helix/random coil to β- sheet. There was no significant difference in the molecular conformation of the SF gels, all of which were mainly composed of β-sheets regardless of the application of shearing, indicating that the gel structure remained stable once formed.
FTIR spectroscopy is a sensitive tool for studying the molecular conformation of SF. The various molecules have their own characteristic absorption bands in the FTIR spectrum. In the absorption spectrum, the position and intensity of amide absorption can reflect the molecular conformation of the SF protein [37]. As seen in Figure 6 spectrum a, the intensities of the absorptionbands at 1631, 1521, 1238 and 700 cm−1 indicate the presence of the β-sheet structure and some α-helix structure in the spontaneously formed ASF gels. There are no significant changes in the spectra of b, c and d, in comparison with spectrum a, indicating that all four gels consist mainly of the β-sheet structure. In Figure 7, the four BSF gels show strong absorption bands at 1635, 1521, 1230 and 700 cm−1, confirming the presence of the β-sheet conformation, which agrees with the XRD data. This result also indicates that the gel structure remains stable once formed.
The results from Figures 1-7 and Tables 1 and 2 show that, for both ASF and BSF, shearing promoted the interand intra-molecular hydrophobic interactions, the rearrangement of hydrogen bonds and the transition from a disordered random coil and/or α-helix structure to a wellordered β-sheet structure, leading to the formation of physically cross-linked gels. With an increasing shearing
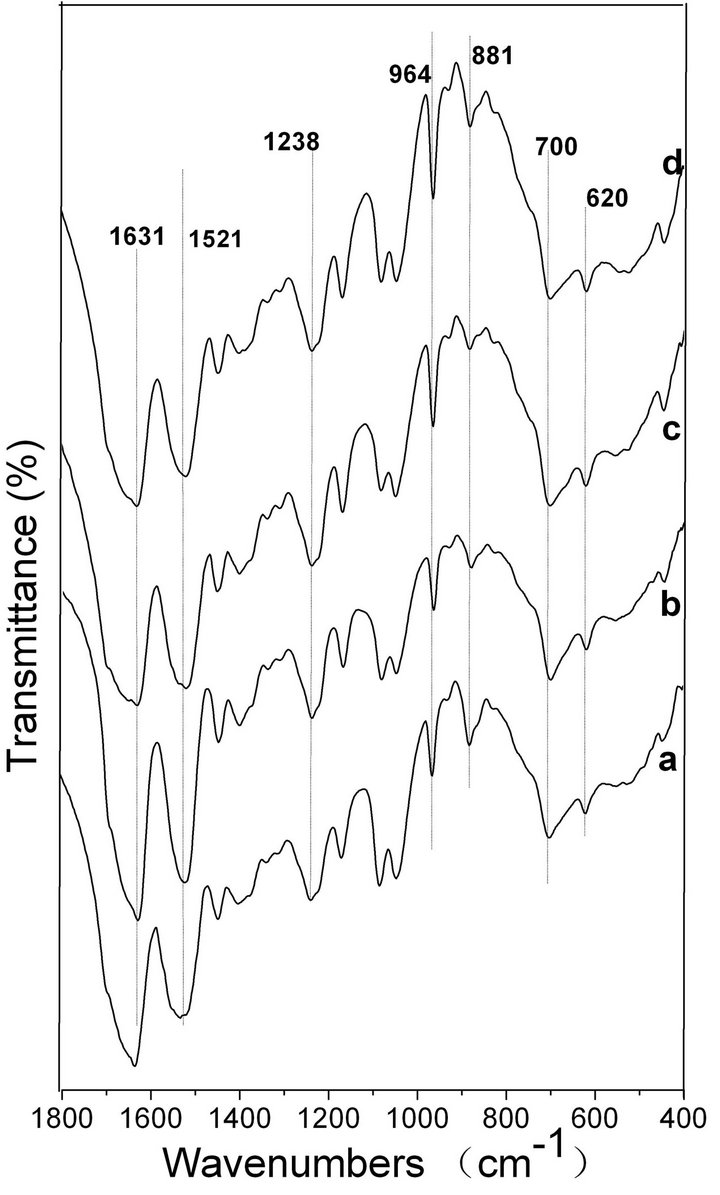
Figure 6. FTIR spectra of the ASF gels induced by shearing for 90 min at rates of 0 rpm (a), 280 rpm (b), 380 rpm (c) and 480 rpm (d).
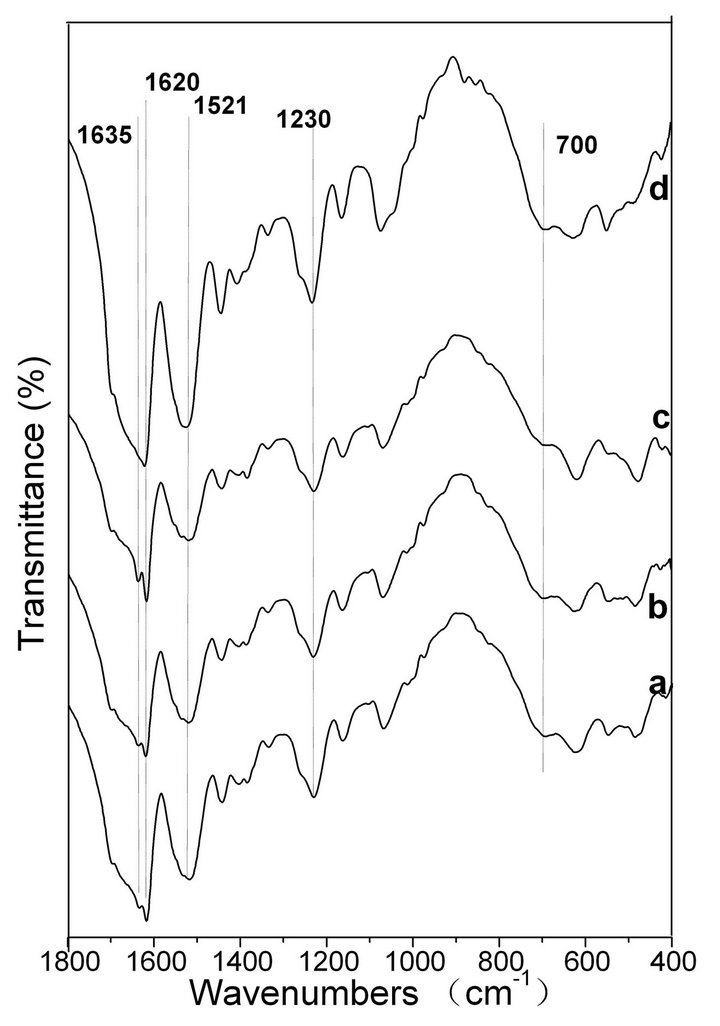
Figure 7. FTIR spectra of the BSF gels induced by shearing for 90 min at rates of 0 rpm (a), 280 rpm (b), 380 rpm (c) and 480 rpm (d).
rate, the physical cross-links among the SF molecules formed more easily. Shearing initiated the formation of β-sheets and accelerated the formation of physical crosslinks, which are responsible for gel stabilization. It can be seen in Figure 1 and Table 2 that the sol-gel transition occurred when the β-sheet content reached nearly 50% which was the minimum content required to maintain the stable hydrogel system in ASF. Hence, the solgel transition rate for SF in solution mainly depends on two factors: the proportion of random coil, α-helix and β-sheet conformers in the initial SF solution and the transition rate from the disordered random coil and/or α-helix structure to a well-ordered β-sheet structure. Accordingly, the key factor for the acceleration of SF gelation is promoting the transition from random coil and/or α-helix to the β-sheet structure.
As shown in Figure 3 and Table 1, the proportion of random coils in the initial BSF solution was nearly 80%, without β-sheet and α-helix structures, which is in agreement with previous reports [38]. The random coil content in the initial ASF solution was approximately 45%, but that of the α-helix was high at 42% in addition to the 12.1% β-sheet fraction, which was quite different from the composition of BSF (Table 1). The secondary structure of a native protein is closely related to its primary structure. It depends on the amino acid sequence and the solution environment in which the protein is dissolved [39,40]. The significant difference in molecular conformation between the initial ASF and BSF aqueous solutions is due to the primary structures. The heavy chain sequence of BSF is characterized by the 94% coverage of Gly-X repeats (where X is Ala at 65%, Ser at 23% and Tyr at 9%) [41]. However, the ASF, except for 155 residues of the amino terminus, is composed of 80 alternate polyalanine-containing units (motifs) [42]. That is, the proportion of Ala residues is greater, while the proportion of Gly residues is less in ASF than in BSF. The Ala is uncharged, and its R-groups are small, so it tends to form the α-helix structure in solution [43,44]. However, the Gly-rich region essentially remains in the random coil state [45]. It is possible that the significant differences in the molecular conformation between ASF and BSF in the initial solution, particularly the higher the α-helix content of ASF, influenced the transition to the β-sheet conformation and accelerated the gelation process. The structure of native SF fiber is known to be the pleated anti-parallel β-sheet structure [46,47]. Hence, the β-sheet conformation is more thermodynamically stable than either the random coil or the α-helix for SF protein. The conformational transition from the random coil and α-helix structures to the β-sheet structure must overcome the energy barrier to form thoroughly stretched peptide chains. The presence of the β-carbon in Ala and other amino acids induces a significant decrease in the conformational entropy of the unfolded state; hence, Gly has greater conformational entropy than Ala [48]. Entropy is the main driving force for the hydrophobic interaction assembly [49]. The conformational entropic cost of transformation from the random coil to the β-sheet structure is larger than the cost of transformation from the α-helix to the β-sheet structure in SF protein.
Mitsuhashi et al. [50] reported that a fibrous “stringlike” crystalline structure was formed when stirring a polyethylene solution. Pennings et al. [51] identified the string-like crystalline structure with electron microscopy, naming it the Shish-kebab structure in 1965. Later studies showed that these structures occurred in elongation, shear and mixed flow conditions, because those conditions essentially promoted polymer stretching along the direction of the force [52].
The results of this study have shown that stirring can accelerate the gelation of SF, especially for ASF (Figure 1). ASF gelation can be controlled within 24 - 48 h. This phenomenon occurs because stirring creates a flowing shearing force that promotes the stretching of the random coil and α-helix chain segments, allowing the reformation of hydrogen bonds and the formation of the β-sheet structure. The primary structures of BSF and ASF have significant differences, so the β-sheet formation is more favorable for ASF. The α-helix content in the initial ASF solution was significantly greater than in BSF under the same conditions (Tables 1 and 2), and the conformational entropic cost of the transition from the α-helix to the β-sheet structure is lower than the cost of the transition from the random coil to the β-sheet structure, leading to the significantly more rapid sol-gel transformation of ASF compared to BSF under the same conditions.
4. Conclusion
It was shown that ASF hydrogels can be successfully and quickly formed using the shearing method without additional chemical processes. Shearing accelerates the solgel transition by inducing rapid physical intraand intermolecular cross-linking via β-sheet formation. When the β-sheet fraction reached nearly 50%, the hydrogels stabilized in the aqueous ASF solution. The ASF gels were prepared quickly (within 24 - 48 h) by stirring the silk fibroin aqueous solutions for 30 - 90 min at defined rates, and the gel structures remained stable once formed. In comparison with BSF, the rapid gelation of ASF through shearing was the result of its alternating polyalaninecontaining units (motifs), which tend to form structures spontaneously, as the entropic cost of the conformational transition from α-helix to β-sheet is less than the cost of the transition from random coil to β-sheet. This controllable method of producing non-chemically cross-linked ASF hydrogels avoids the complications and inherent toxicity to cells or cytokines associated with some unpurified or remaining cross-linking additives, and it may therefore provide a potential method for the production of silk-based tissue engineering scaffolds and drug/ growth delivery carriers.
5. Acknowledgements
This work was supported by the College Natural Science Research Project of Jiangsu Province (12KJA430003), the National Nature Science Foundation of China (309 70714), the Key Laboratory Opening Program of Jiangsu Higher Education Institutions (KJS1216) and the Priority Academic Program Development of Jiangsu Higher Education Institutions.
REFERENCES
- G. R. Plaza, P. Corsini, E. Marsano, J. Perez-Rigueiro, M. Elices, C. Riekel, C. Vendrely and G. V. Guinea, “Correlation between Processing Conditions, Microstructure and Mechanical Behavior in Regenerated Silkworm Silk Fibers,” Journal of Polymer Science Part B: Polymer Physics, Vol. 50, No. 7, 2012, pp. 455-465. doi:10.1002/polb.23025
- A. S. Lammel, X. Hu, S. H. Park, D. L. Kaplan and T. R. Scheibel, “Controlling Silk Fibroin Particle Features for Drug Delivery,” Biomaterials, Vol. 31, No. 16, 2010, pp. 4583-4591. doi:10.1016/j.biomaterials.2010.02.024
- M. Z. Li, M. Ögiso and N. Minoura, “Enzymatic Degradation Behavior of Porous Silk Fibroin Sheets,” Biomaterials, Vol. 24, No. 2, 2003, pp. 357-365.
- R. E. Unger, S. Ghanaati, C. Orth, A. Sartoris, M. Barbeck, S. Halstenberg, A. Motta, C. Migliaresi and C. J. Kirkpatrick, “The Rapid Anastomosis between Prevascularized Networks on Silk Fibroin Scaffolds Generated in Vitro with Cocultures of Human Microvascular Endothelial and Osteoblast Cells and the Host Vasculature,” Biomaterials, Vol. 31, No. 27, 2010, pp. 6959-6967. doi:10.1016/j.biomaterials.2010.05.057
- R. Okabayashi, M. Nakamura, T. Okabayashi, Y. Tanaka, A. Nagai and K. Yamashita, “Efficacy of Polarized Hydroxyapatite and Silk Fibroin Composite Dressing Gel on Epidermal Recovery from Full-Thickness Skin Wounds,” Journal of Biomedical Materials Research Part B: Applied Biomaterials, Vol. 90B, No. 2, 2009, pp. 641-646.
- B. B. Mandal and S. C. Kundu, “Cell Proliferation and Migration in Silk Fibroin 3D Scaffolds,” Biomaterials, Vol. 30, No. 15, 2009, pp. 2956-2965. doi:10.1016/j.biomaterials.2009.02.006
- S. Sahoo, S. L. Tohb and J. C. H. Goha, “A bFGF-Releasing Silk/PLGA-Based Biohybrid Scaffold for Ligament/Tendon Tissue Engineering Using Mesenchymal Progenitor Cells,” Biomaterials, Vol. 31, No. 11, 2010, pp. 2990-2998. doi:10.1016/j.biomaterials.2010.01.004
- S. Madduri, M. Papaloïzos and B. Gander, “Trophically and Topographically Functionalized Silk Fibroin Nerve Conduits for Guided Peripheral Nerve Regeneration,” Biomaterials, Vol. 31, No. 8, 2010, pp. 2323-2334. doi:10.1016/j.biomaterials.2009.11.073
- P. W. Madden, J. N. X. Lai, K. A. George, T. Giovenco, D. G. Harkin and T. V. Chirila, “Human Corneal Endothelial Cell Growth on a Silk Fibroin Membrane,” Biomaterials, Vol. 32, No. 17, 2011, pp. 4076-4084. doi:10.1016/j.biomaterials.2010.12.034
- E. Wenk, H. P. Merkle, L. Meinel, “Silk Fibroin as a Vehicle for Drug Delivery Applications,” Journal of Controlled Release, Vol. 150, No. 2, 2011, pp. 128-141. doi:10.1016/j.jconrel.2010.11.007
- E. Ruoslahti and M. D. Pierschbacher, “New Perspectives in Cell Adhesion: RGD and Integrins,” Science, Vol. 238, No. 4826, 1987, pp. 491-497. doi:10.1126/science.2821619
- A. J. Meinel, K. E. Kubow, E. Klotzsch, M. GarciaFuentes, M. L. Smith, V. Vogel, H. P. Merkle and L. Meinel, “Optimization Strategies for Electrospun Silk Fibroin Tissue Engineering Scaffolds,” Biomaterials, Vol. 30, No. 17, 2009, pp. 3058-3067. doi:10.1016/j.biomaterials.2009.01.054
- C. Acharya, S. K. Ghosh and S. C. Kundu, “Silk Fibroin Film from Non-Mulberry Tropical Tasar Silkworms: A Novel Substrate for in Vitro Fibroblast Culture,” Acta Biomaterialia, Vol. 5, No. 1, 2009, pp. 429-437. doi:10.1016/j.actbio.2008.07.003
- Y. Z. Wang, D. D. Rudym, A. Walsh, L. Abrahamsen, H. J. Kim, H. S. Kim, C. Kirker-Head and D. L. Kaplan, “In Vivo Degradation of Three-Dimensional Silk Fibroin Scaffolds,” Biomaterials, Vol. 29, No.24-25, 2008, pp. 3415- 3428. doi:0.1016/j.biomaterials.2008.05.002
- M. Z. Li, W. Tao, S. Z. Lu and C. X. Zhao, “Porous 3-D Scaffolds from Regenerated Antheraea pernyi Silk Fibroin,” Polymers for Advanced Technologies, Vol. 19, No. 3, 2008, pp. 207-212. doi:10.1002/pat.998
- C. Patra, S. Talukdar, T. Novoyatleva, S. R. Velagala, C. Mühlfeld, B. Kundu, S. C. Kundu and F. B. Engel, “Silk Protein Fibroin from Antheraea mylitta for Cardiac Tissue Engineering,” Biomaterials, Vol. 33, No. 9, 2012, pp. 2673-2680. doi:10.1016/j.biomaterials.2011.12.036
- E. S. Gil, R. J. Spontak and S. M. Hudson, “Effect of β- Sheet Crystals on the Thermal and Rheological Behavior of Protein-Based Hydrogels Derived from Gelatin and Silk Fibroin,” Macromolecular Bioscience, Vol.5, No. 8, 2005, pp. 702-709. doi:10.1002/mabi.200500076
- M. N. Collins and C. Birkinshaw, “Morphology of Crosslinked Hyaluronic Acid Porous Hydrogels,” Journal of Applied Polymer Science, Vol. 120, No. 2, 2011, pp. 1040-1049. doi:10.1002/app.33241
- M. L. Floren, S. Spilimbergo, A. Motta and C. Migliaresi, “Carbon Dioxide Induced Silk Protein Gelation for Biomedical Applications,” Biomacromolecules, Vol. 13, No. 7, 2012, pp. 2060-2072. doi:10.1021/bm300450a
- N. Guziewicz, A. Best, B. Perez-Ramirez and D. L. Kaplan, “Lyophilized Silk Fibroin Hydrogels for the Sustained Local Delivery of Therapeutic Monoclonal Antibodies,” Biomaterials, Vol. 32, No. 10, 2011, pp. 2642- 2650.
- N. Bhattarai, J. Gunn and M. Zhang, “Chitosan-Based Hydrogels for Controlled, Localized Drug Delivery,” Advanced Drug Delivery Reviews, Vol. 62, No. 1, 2010, pp. 83-99. doi:10.1016/j.addr.2009.07.019
- X. Hu, Q. Lu, L. Sun, P. Cebe, X. Q. Wang, X. H. Zhang and D. L. Kaplan, “Biomaterials from UltrasonicationInduced Silk Fibroin-Hyaluronic Acid Hydrogels,” Biomacromolecules, Vol. 11, No. 11, 2010, pp. 3178-3188. doi:10.1021/bm1010504
- T. Yucel, P. Cebel and D. L. Kaplan, “Vortex-Induced Injectable Silk Fibroin Hydrogels,” Biophysical Journal, Vol. 97, No. 7, 2009, pp. 2044-2050. doi:10.1016/j.bpj.2009.07.028
- A. Matsumoto, J. Chen, A. L. Collette, U. J. Kim, G. H. Altman, P. Cebe and D. L. Kaplan, “Mechanisms of Silk Fibroin Sol-Gel Transitions,” The Journal of Physical Chemistry B, Vol. 110, No. 43, 2006, pp. 21630-21638. doi:10.1021/jp056350v
- U. J. Kim, J. Park, C. M. Li, H. J. Jin, R. Valluzzi and D. L. Kaplan, “Structure and Properties of Silk Hydrogels,” Biomacromolecules, Vol. 5, No. 3, 2004, pp. 786-792. doi:10.1021/bm0345460
- X. Q. Wang, J. A. Kluge, G. G. Leisk and D. L. Kaplan, “Sonication-Induced Gelation of Silk fibroin for Cell Encapsulation,” Biomaterials, Vol. 29, No. 8, 2008, pp. 1054- 1064. doi:10.1016/j.biomaterials.2007.11.003
- K. Numata, T. Katashima and T. Sakai, “State of Water, Molecular Structure, and Cytotoxicity of Silk Hydrogels,” Biomacromolecules, Vol. 12, No. 6, 2011, pp. 2137- 2144.
- E. S. Gil, D. J. Frankowski, R. J. Spontak and S. M. Hudson, “Swelling Behavior and Morphological Evolution of Mixed Gelatin/Silk Fibroin Hydrogels,” Biomacromolecules, Vol. 6, No. 6, 2005, pp. 3079-3087. doi:10.1021/bm050396c
- M. Fini, A. Mott, P. Torricelli, G. Giavaresi, A. N. Nicoli, M. Tschon, R. Giardino and C. Migliaresi, “The Healing of Confined Critical Size Cancellous Defects in the Presence of Silk Fibroin Hydrogel,” Biomaterials, Vol. 26, No. 17, 2005, pp. 3527-3536. doi:10.1016/j.biomaterials.2004.09.040
- X. G. Li, L.Y. Wu, M. R. Huang, H. L. Shao and X. C. Hu, “Conformational Transition and Liquid Crystalline State of Regenerated Silk Fibroin in Water,” Biopolymers, Vol. 89, No. 6, 2008, pp. 497-505. doi:10.1002/bip.20905
- S. Q. Yan, C. X. Zhao, X. F. Wu, Q. Zhang and M. Z. Li, “Gelation Behavior of Antheraea pernyi Silk Fibroin,” Science China Chemistry, Vol. 53, No. 3, 2010, pp. 535- 541. doi:10.1007/s11426-010-0093-0
- S. Brahms, “Determination of Protein Secondary Structure in Solution by Vacuum Ultraviolet Circular Dichroism,” Journal of Molecular Biology, Vol. 138, No. 2, 1980, pp. 149-178. doi:10.1016/0022-2836(80)90282-X
- N. J. Greenfield, “Using Circular Dichroism Spectra to Estimate Protein Secondary Structure,” Nature Protocols, Vol. 1, 2007, pp. 2876-2890. doi:10.1038/nprot.2006.202
- W. Tao, M. Z. Li and C. X. Zhao, “Structure and Properties of Regenerated Antheraea pernyi Silk Fibroin in Aqueous Solution,” International Journal of Biological Macromolecules, Vol. 40, No. 5, 2007, pp. 472-478. doi:10.1016/j.ijbiomac.2006.11.006
- J. X. He, Y. M. Cheng and S. Z. Cui, “Preparation and Characterization of Electrospun Antheraea pernyi Silk Fibroin Nanofibers from Aqueous Solution,” Journal of Applied Polymer Science, Vol. 128, No. 2, 2013, pp. 1081- 1088. doi:10.1002/app.38233
- K. H. Zhang, Q. Z. Yu and X. M. Mo, “Fabrication and Intermolecular Interactions of Silk Fibroin/Hydroxybutyl Chitosan Blended Nanofibers,” International Journal of Molecular Sciences, Vol. 12, 2011, No. 4, pp. 2187-2199. doi:10.3390/ijms12042187
- B. B. Mandal, S. Kapoor and S. C. Kundu, “Silk Fibroin/Polyacrylamide Semi-Interpenetrating Network Hydrogels for Controlled Drug Release,” Biomaterials, Vol. 30, No. 14, 2009, pp. 2826-2836. doi:10.1016/j.biomaterials.2009.01.040
- S. W. Ha, A. E. Tonelli and S. M. Hudson, “Structural Studies of Bombyx mori Silk Fibroin during Regeneration from Solutions and Wet Fiber Spinning,” Biomacromolecules, Vol. 6, No. 3, 2005, pp. 1722-1731. doi:10.1021/bm050010y
- F. U. Hart and M. Hayer-Hart, “Converging Concepts of Protein Folding in Vitro and in Vivo,” Nature Structural & Molecular Biology, Vol. 16, No. 6, 2009, pp. 574-581. doi:10.1038/nsmb.1591
- J. R. Macdonald and J. R. Johnson, “Environmental Features are Important in Determining Protein Secondary Structure,” Protein Science, Vol. 10, No. 6, 2001, pp. 1172-1177. doi:10.1110/ps.420101
- C. Vepari and D. L. Kaplan, “Silk as a Biomaterial,” Progress in Polymer Science, Vol. 32, No. 8-9, 2007, pp. 991-1007. doi:10.1016/j.progpolymsci.2007.05.013
- J. G. Hardy and T. R. Scheibel, “Silk-Inspired Polymers and Proteins,” Biochemical Society Transactions, Vol. 37, No. 4, 2009, pp. 677-681. doi:10.1042/BST0370677
- M. Blaber, X. J. Zhang and B. W. Matthews, “Structural Basis of amino Acid Alpha Helix Propensity,” Science, Vol. 260, No. 5114, 1993, pp. 1637-1640. doi:10.1126/science.8503008
- S. M. LaPointe, S. Farrag, H. J. Bohorquez and R. J. Boyd, “QTAIM Study of an α-Helix Hydrogen Bond Network,” The Journal of Physical Chemistry B, Vol. 113, No. 31, 2009, pp. 10957-10964. doi:10.1021/jp903635h
- Y. Nakazawa and T. Asakura, “Heterogeneous Exchange Behavior of Samia Cynthia Ricini Silk Fibroin during Helix-Coil Transition Studied with 13C NMR,” FEBS Letters, Vol. 529, No. 2-3, 2002, pp. 188-192. doi:10.1016/S0014-5793(02)03332-X
- S. Nagarkar, A. Patil, A. Lele, S. Bhat, J. Bellare and R. A. Mashelkar, “Some Mechanistic Insights into the Gelation of Regenerated Silk Fibroin Sol,” Industrial & Engineering Chemistry Research, Vol. 48, No. 17, 2009, pp. 8014-8023. doi:10.1021/ie801723f
- C. S. Ki, Y. H. Park and H. J. Jin, “Silk Protein as a Fascinating Biomedical Polymer: Structural Fundamentals and Applications,” Macromolecular Research, Vol. 17, No. 12, 2009, pp. 935-942. doi:10.1007/BF03218639
- K. A. Scott, D. O. V. Alonso, S. Sato, A. R. Fersht and V. Daggett, “Conformational Entropy of Alanine versus Glycine in Protein Denatured States,” PNAS, Vol. 104, No. 8, 2007, pp. 2661-2666. doi:10.1073/pnas.0611182104
- D. Chandler, “Interfaces and the Driving Force of Hydrophobic Assembly,” Nature, Vol. 437, No. 7059, 2005, pp. 640-647. doi:10.1038/nature04162
- S. Mitsuhashi, M. Morimura, K. Kono and H. Oshima, “Elimination of Drug Resistance of Staphylococcus aureus By Treatment with Acriflavine,” Journal of Bacteriology, Vol. 86, No. 1, 1963, pp. 162-164.
- A. J. Penning and A. M. Kiel, “Fractionation of Polymers by Crystallization from Solution, III. On the Morphology of Fibrillar Polyethylene Crystals Grown in Solution,” Colloid & Polymer Science, Vol. 205, No. 2, 1965, pp. 160-162. doi:10.1007/BF01507982
- A. J. Pennings, Vander, J. M. A. A. Mark and A. M. Kiel, “Hydrodynamically Induced Crystallization of Polymers from Solution,” Colloid and Polymer Science, Vol. 237, No. 2, 1970, pp. 336-358.
NOTES
*Corresponding author.